Endy:Screening plasmid/Terminator characterization/Protocols
This is a work in progress, protocol is currently incomplete
Purpose
Characterize the in vivo termination efficiency of a Biobricked terminator quickly and reliably.
Background
The terminators should be present in one of the PoPS based screening plasmids, this protocol refers to characterization of terminators in Screening Plasmid 1.0 in CW2553+pJat8. pJat8 provides Gen resistance and the screening plasmid has Amp resistance.
The current characterization protocol follows the following steps:
Day 1:
- Grow the cells overnight in a rich media to achieve density.
Day 2:
- Dilute back to return the cells to mid-log in M9/gly
- Innoculate experimental cultures of M9/gly/ara
Day 3:
- Harvest cells for FACS
- Run FACS
Procedure
Each culture has a different growth rate, so ensuring that the intermediate dilution and experimental culture remain in log phase can be difficult. Additionally, the final culture must match induction time (so that the fluorophore concentration reaches steady state) and growth time (so that the culture is dense enough to measure at the end of the experiment, but still in mid-log). Rough guidelines for timing are included below.
Day 1
Set up Overnights
It is only necessary to set up one 5ml overnight culture per experiment device (terminator) or control. These cells typically grow very slowly. I set up overnights early in the afternoon in a richer media (M9/glucose works reasonably well) and give them plenty of time to grow up.
- Controls
- Negative Control - CW2553/pJat8
- Empty Plasmid - This provides a baseline for calibrating GFP and RFP
- GFP only - This enables quantification of bleed from GFP in RFP filter
- (We have these results should link to the measurements here.)
Day 2
Dilute back in AM
For high copy plasmids, I estimate a doubling time of 1.5 to 2 hours. I generally dilute back 100x into 5ml of M9/glycerol and grow for 8-10 hours. For low copy plasmids, either shorten the growth time (5-6 hours rather than 8-10) or increase the dilution.
Set up overnight experimental cultures
A 12 hours induction seems to provide sufficient time for the fluorophores to reach steady state (previous results with an empty screening plasmid have shown unchanged levels between 12 and 14 hours). For a high copy plasmid, innoculating at OD 0.002 works reasonably well. Lower copy plasmids will need smaller innoculums (OD 0.0001-0.0005).
In previous experiments have used 3 arabinose induction levels to test terminators, all of them in the 'high' range of induction: (0.0003%, 0.0015%, and 0.003%). It is usually a good idea to make up large batches of media + arabinose and then make 5ml aliquots of this for each experimental device -- this ensures they all see the same levels of arabinose. Each experimental strain as well as the empty plasmid control should be innoculated in all 3 arabinose concentrations, the negative control can be grown with no arabinose, and the GFP only control should be grown in 0.003% to maximize the FL output. So total samples will be ((EXP+1)*#arab concentrations + 2) * #replicates
Day 3
Prepare FACS Samples
Take 1 ml aliquots and place on ice.
Run samples on FACS
- Print this sheet to take along to the FACS facility.
- Set PMT voltages based on the Empty plasmid control at full induction. The GFP and RFP signals should both be centered around 100 FLunits.
- The goal here is to take advantage of the full range of outputs from the device to get the best signal. It may make more sense in the future to move to higher PMT settings, since theoretically full induction of the empty plasmid control should be the highest readings we see in a terminator characteriation experiment (baring termiantors serving as promoters). However, apparently a voltage of <680 is prefered due to issues with the MOFLO (ask about how firm a limit this is).
- The droplet maker can be turned off to reduce noise in the signal if you are not sorting the cells.
- Run beads (~6 drops in 500ml) - this is used to verify that the lasers are aligned as well as to serve as a calibration between runs, you can read more about this in the analysis techniques. Be sure to save the bead calibration data.
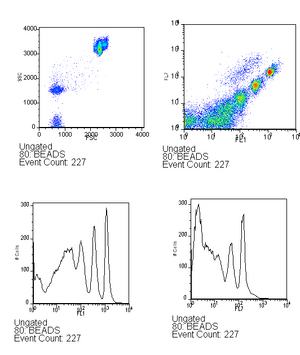
Beads should look approximately like this with good alignment. - Run the empty plasmid again, followed by the negative control.
- This allows you to determine the "noise region", since you shouldn't trust readings that are within the range of autoflourescence of your cells. Also, it makes sure that the method being used to clear the tubing between samples is working well. If you still see many positive cells in the negative control then you can try running bleach between samples. (This is maybe more important between different terminators then between different arabinose concentrations).
- Run the experimental samples. (have been collecting ~30K cells / sample)

Analysis
Contact
- protocol based on screening plasmid protocols developed by Josh Michener
