CH391L/S2013 Taylor Pursell March 20 2013
Identification of Early Replicating Fragile Sites that Contribute to Genome Instability. Barlow JH, Faryabi RB, Callén E, Wong N, Malhowski A, Chen HT, Gutierrez-Cruz G, Sun HW, McKinnon P, Wright G, Casellas R, Robbiani DF, Staudt L, Fernandez-Capetillo O, Nussenzweig A. Cell. 2013 Jan 22. pii: S0092-8674(13)00008-1.
Background
B-Lymphocytes
B lymphocytes, or B-cells, originate in the bone marrow. The B-cells are then circulated in the blood and become activated through interactions with an antigen and/or T-cells. In this case we will focus on T-cell dependent activation where B-cells undergo terminal maturation after becoming activated through interaction with an antigen and a T-cell. Once activated, large B-cells undergo rapid proliferation and genomic rearrangements important for expansion and specification of the antibody repertoire [1,4,5]. These genome changes come in two classes:
- Somatic Hypermutationi(SHM) in which point mutations, or more rarely deletion or insertions, are induced in the variable region of immunoglobulin (Ig) genes and their surrounding regions.
- Class Switch Recombination (CSR) where one constant region of Ig is deleted and replaced with another[1,5].
Activation-Induced Cytidine Deaminase and DNA Damage
An enzyme called activation-induced cytidine deaminase (AID) is required for SHM and CSR to occur [5,6]. By deaminating cytosine residues on ssDNA that are exposed during Ig transcription, the enzyme turns the cytosine residue into a uridine. The mismatched uridine is glycosylated and removed by uracil DNA glycosylase (UNG) allowing the apyrimidic endonuclease (APE) to generate the DNA breaks needed for SHM and CSR to occur [5].
AID has been shown to act in the presence of a particular motif: DGYW (D= any bas but C ; G= guainine; Y= pyrimidine ; W= adenosine or thymidine) [6]. Although this is its primary function, it is not uncommon for it to act on other DNA segments containing motif including oncogenes like c-Myc [1].
Replication and Fragile Sites
The eukaryotic genome is divide into regions which are programmed to replicate at different times during S-phase of the cell cycle. There are two groups of replication origins: early firing origins and late origins. Origins near active genes, or genes whose products are important to the cell, fire in the first half of S-phase in a process called early replication. In contrast, origins near inactive genes fire during the second half of S-phase and are associated with late replication[2].
Chromosomal fragile sites are specific stretched of genetic material that are prone to gaps and breaks following replication leading to chromosome instability; these fragile sites are put into two classes: rare and common fragile sites. Rare fragile sites are those found in only a few individuals and are usually associated with genetic disorders while common fragile sites (CFS) are found in all individuals [3]. Common fragile sites are can be associated with large genes, long stretches of AT rich regions, and stalled DNA replication [1,3]. Common fragile sites are associated with late replicating origins [1,2,3].
Introduction
In studies preformed in yeast, origins at which replication fork collaspe occurs in response to stalled replication have been identified [7]. These origins do not line up with "replication slow zones" or the yeast equivalent of CFS found at late replication origins [8,9] suggesting there is another class of fragile sites in yeast.
The researchers in this study identify similar early replicating origins at which DNA lesions are found in B-cells; they termed these sites early replicating fragile sites (ERFSs) [1].
Methods
ChIP and ChIP-Seq

Mouse splenic B-cell samples were stimulated with LSP/IL4 to cause the cells to synchronously enter cell cycle. After 22 hours, cells should be in S phase (monitored by FACs). Cells were then treated with 10 mM hydroxyurea (HU) which arrests cells at G1/S phase and then cells were harvested. Cells were crosslinked in order to ensure any protein bound to DNA would remain bound. Cells are sonicated in order to break up cells and to fragment DNA. Sonicated chromatin samples are then incubated with an antibody specific to the protein of interest (e.g. RPA, BRCA1, etc.) and magnetic breads specific to the constant region of the antibody used. These magnetic beads are used to pull out or precipitate the anitbody, protein, and bound sequence of DNA. The protein and DNA can be separated from the bead and prepared for sequencing. For schematic see figure to the right from the manufacture's website (http://www.illumina.com/technology/chip_seq_assay.ilmn).
Once genetic material is harvested, it was prepared for micro sequencing using Illumina’s protocol for sample preparation and was analyzed using the Genome Analyzer IIx.

Fluorescent in-situ hybridization (FISH) analysis
Fluorescent probes that bind to complementary sequences on chromosomes are used to visualize abnormalities. Fluorescent microscopy can be used to find where the probe is bound. In this paper, blue is DAPI-stained DNA, green is the insert in the BAC (either ERFSs or CFSs) and red is telomeric DNA.
Results
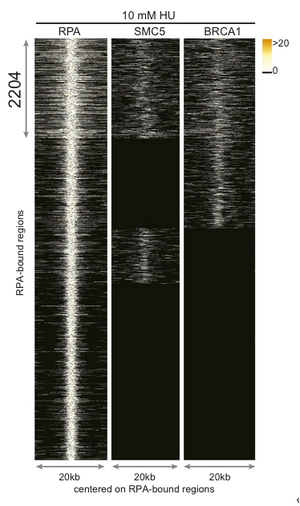
First, researchers needed to map early replication origins in an activated B-cell genome. To do this they performing ChIP with anti-replication protein A (RPA). They harvested fresh mouse splenic B-cells, stimulated them with LPS/IL4 to enter cell cycle. They then inhibited replication in G1/S phase using hydroxyurea (HU) allowing RPA binds to ssDNA at stalled forks [1]. Treatment with HU serves to simulate the stalling of replication that might naturally occur in rapidly dividing B-cells after activation.
In addition to RPA, the researchers looked for the localization of recombination proteins BRCA1 and SMC5 to regions also occupied by RPA in order to identify regions in which the fork was not only stalled but had collapsed and required homologous recombination repair. From this ChIP data, they were able to identify 2204 regions were RPA, BRCA1, and SMC5 were all localized. Upon further characterization of the 2204 regions, it was found that they contained repetitive elements which are known fork barriers [1]. These regions were also significantly more GC rich compared to the rest of the genome; this characteristic further differentiates them from CFSs which are AT rich regions [1,3]. The researchers termed these regions early replicating fragile sites or ERFSs.Because most (80%) of ERFSs are within 300 kb of one another, researchers grouped the ERFSs into regions they call ERFS hot spots. They then narrowed the hotspots down to those with a ChIP footprint larger then 10kb to give 619 hotspots scattered throughout the genome (Figure B, below) with the notable exception of the sex chromosomes. The top 15 hotspots with the highest RPA/BRAC1/SMC5-binding strength were analyzed further to reveal that 8 out of 15 rearranged in B-cell lymphoma [1].
Next, they visualized the chromosome damage, or abbreviations, by arresting replication in G1/S phase wildtype B-cells and B-cells unable to undergo homologous recombination (XRCC2-/-) with HU and then examining metaphase chromosome spreads with FISH analysis. They probed for ERFSs (MHCII, GIMAP, SWAP70, BACH2, IKZF1, and FOXP1), control regions (CNTNAP4 and SLITRK6), and two CFSs (FRA8E1 and FRA14A2). The regional probes are in green, the blue is DAPI stained chromosomes, and red is a probe for telomeric regions. While DNA lesions were induces at ERFSs, not CRFs, when cell replication was stalled in early S-phase (Figure A, above), the researchers were also able to induce breaks at CFSs only by completely inhibiting replication with aphidicolin overnight. These results confirm that damage arising at ERFS and CRF are the result of two separate processes: arise from fork collapse during early replication and failure to replicate, respectively.
Next, the researchers looked for conditions which effect the fragility of the ERFSs. They found that inhibition of ATR kinase, which protects the against breaks at CFSs in addition to stabilizing stalled forks and assisting in fork restart, causes damage at CFSs and ERFSs. In addition to ATR kinase inhibition, increased transcriptional activity at ERFSs was found to increase fragility of the site. Another concern is how oncogenic stress effects fragile sites. To test the effect of oncogenic stress on the fragility of ERFSs, researchers over expressed c-myc (an target of extranious AID activation). c-myc has been implicated in replication initiation and origin firing in B-cells. The researchers overexpressed c-myc in cells that could not under homologous recombination (XRCC-/-) and used FISH metaphase spreads to look for chromosome abnormalities. This overexpression correlated to an increase in breaks at ERFSs and CFSs.
Another important characteristic of ERFSs is that damage occurring at these sites is AID-independent. To test this, researchers used a AID-/- B-cells, or cell lacking AID. By treating both AID-/- and WT B-cells with LPS/IL4 in combination with ATRi, researchers were able to combine replicative stress in addition to AID-dependent damage. Under these conditions, researchers found that Ig abbreviations were abundant in WT cells and absent in AID-/- cell indicating damage occurring at the Ig locus was AID-dependent. In contrast, breakage at ERFSs remain relatively unchanged at most sites (Figure 5c, below). This data suggests that breakage at ERFSs is AID-independent.

The researchers then moved into human cancer models. By overlaying the mouse ERFSs with human genome using 2 different approached, they were able to generate 2205 synthetic ERFS regions in humans which would correlate to the ERFSs identified in mice. They then compared these regions to 190 minimal common regions (MCRs) identified using 203 diffuse large B-cell lymphoma (DLBCL) biopsies or regions which had gain or loss of genetic material. They found that 450 ERFS coincided with 98 MCRs. They quantified this overlap comparing it to an expectation of overlap generated using a permutation model, but also to CFS overlap. They say a 20% enrichment over thei expectation and 80% relative to CFSs. In addition to MCRs, they also looked for cancer genes associated with ERFS and found that out of 64 punative cancer genes [published by Bignell et al. 2010] roughly 39% were associated with ERFS.
Based on data collected researchers generated a model for how off-target AID activity in combination with breaks occurring at ERFSs might lead to cancer. Their model describes are two possible ways to get to a potentially cancerous genetic mutation. The first involves AID off-target induction of a DSB at an oncogene in G1 phase; this DSB remains into S phase. In S phase, replication stress causes a break at an ERFS containing an oncogene. Under the correct conditions, this will result in a translocation. The second involves an unresolved fork collapse and subsequence DSB at an ERFSs oncogene which is carried through cell division. Upon entering G1 phase, AID dependent damage occurs allowing for translocation.
Significance/Applications
The paper challenges the assumption that replication related genome instability is associated with a certain set of characteristics found in CFSs such as AT-enrichment, low origin density, late replication, and dense chromosome structure. The researchers are able to provide data which suggests that the other extreme (GC-richness, high origin and gene density, early replication, and high transcriptional activity) can also lead to genome instability.
In relation to cancer genetics, this paper presents a method which could be used to identify the cellular origin of a cancer. Because transcriptional activity and replication timing differ across cell types, each cell type will have its own set of ERFSs. By looking for abnormalities at ERFSs of different cell types, a physician can identify the cellular origin and cause of the cancer.
References
- Barlow, J. H. et al. Identification of Early Replicating Fragile Sites that Contribute to Genome Instability. Cell 152:1-13 (2013)
- Goren A, Cedar H. Replicating by the clock. Nature Reviews Molecular Cell Biology In Press 4: 25-32 (2003)
- Durkin, S. G. & Glover, T. W. Chromosome Fraglie Sites. Annual Reviews Genetic 41: 169-192 (2007)
- Parham, Peter. The Immune System. Garland Science, 2. (2005)
- Durandy, A. Activation-induced cytidine deaminase: a dual role in class-switch recombination and somatic hypermutation. Eur. J. Immunol 33: 2069-2073 (2003)
- Kenter, A. L. & Bhattacharya, P. AID: a very old motif newly recognized. Nature Immunology 5: 1203-04 (2004)
