Biomod/2014/OhioMOD/experimentnotes
<html> <head> <script src="http://ajax.googleapis.com/ajax/libs/jquery/1.11.1/jquery.min.js"></script> <script src="http://maxcdn.bootstrapcdn.com/bootstrap/3.2.0/js/bootstrap.min.js"></script> <link href="http://maxcdn.bootstrapcdn.com/bootswatch/3.2.0/yeti/bootstrap.min.css" rel="stylesheet"> <style>
- content{margin-left:0;}
.navbar{ top:8.5px; position:fixed; width:96.8%; } /*clearing the side openwetware panels*/ .firstHeading, #column-one, #p-bookmarks, #p-history , .portlet{ display:none; }
- bodyContent/*clearing the side openwetware panels
.firstHeading, #column-one, #p-bookmarks, #p-history , .portlet{ display:none; }
- content#bodyContent, {
/*display:none;*/ background-color:#FFFFFF; }
- OSUfooter{
clear:both; }
- footer{display: none;}
/*Done clearing*/
</style>
</head>
</html> <html> <head> <style> center {padding-bottom:50px;}
- ExperimentNotes{
width: 700px ; margin-left: auto ; margin-right: auto ; margin-bottom: 50px;
}
- list2
{
text-indent:50px;
}
/*fixed icon code start*/
- fixedbutton {
position: fixed; bottom: 53px; right: 10px;
} /*fixed icon code end*/
</style>
<style type="text/css"> p{text-indent:50px;} </style>
</head> <body>
Experiment Notes
| Table of Contents | |
|---|---|
|
<A HREF="#scroll1">1. Cellular Uptake Experiment</A>
|
|
1. Cellular Uptake Experiment
Treatments:
1. Cells only: Lysotracker stained cells only.
2. Branch (Br scr): Lysotracker stained cells with TOPRO3 stained branch scrambled overhang.
3. Block O (BO scr): Lysotracker stained cells with TOPRO3 stained Block O scrambled overhangs.
Procedure:
1. Staining of structures
1.1 Measure the concentration of structures and concentration of dye needed to stain 50 uL of structures
1.2 Add correct amount of dye to 50 uL of structure and incubate overnight
2. Staining of cells
2.1 Combine 0.1 uL of 1 mM Lysotracker with 1 mL of Human RPMI media to get final concentration of 100 nM. Incubate at 37 C.
2.2 Count cells. Aliquot 3 samples with ~75,000 cells in each.
2.3 Wash cells with PBS. Resuspend in 200 uL of human media containing Lysotracker dye. Incubate for 1.5 hrs.
3. Structure addition
3.1 After incubation, wash cells with PBS
3.2 Resuspend cells in clear media with 10% FBS.
a 175 uL clear media and 25 uL FBS.
3.3 Plate cells in 8 well imaging plate. Add 50 uL of structures to corresponding plates.
a. Sample 1 will be unstained cells only (CO
Add 50 uL of TAE buffer
b. Sample 2 will be stained cells with branch stained with TOPRO.
structure concentration: 0.2 nM
c. Sample 3 will be stained cells with Block O stained with TOPRO.
structure concentration: 0.2 nM
4. TIRF imaging
4.1 Immediately image under TIRF to get t=0 data. After imaging, reincubate for 4 hrs
Results:
t = 0
• Cells only:
<figure>
<img src="http://openwetware.org/images/4/48/Exptfig1.png"height="217" width="624"/>
<figcaption>Figure 1: Cells stained with Lysotracker only.</figcaption>
</figure>
The cells only images at time 0 showed ideal Lysotracker staining, with very little fluorescent signal in the 640 channel.
• Branch
<figure>
<img src="http://openwetware.org/images/9/9c/Expfig2.png"height="209" width="624"/>
<figcaption>Figure 2: Stained cells incubated with stained branch structures.</figcaption>
</figure>
</brThe images of cells with branch structures exhibits a strong signal in both the 488 and the 640 channel, confirming the successful staining by both the Lysotracker and the TOPRO3. However, while the Lysotracker signal is present in both cells, the TOPRO3 signal is present only in the cell with a highly granular interior, suggesting a cell nearing death.
• Block O
<figure>
<img src="http://openwetware.org/images/f/fd/Expfig3.png"height="206" width="624"/>
<figcaption>Figure 3: Stained cells with stained Block O structures.</figcaption>
</figure>
The cells incubated with Block O showed a weaker Lysotracker staining than the cells with branch. However, the stain was still discernible. As in the branch sample, there was a strong fluorescent signal in the 640 channel which corresponds to stained structures. However, as with the branch sample, the fluorescence originates from dead cell debris rather than any live cells.
There was no detectable co-localization of the two signals in any of the samples imaged at t = 0.
t = 4
• Cells only
<figure>
<img src="http://openwetware.org/images/b/bb/Expfig4.png"height="206" width="620"/>
<figcaption>Figure 4: Cells stained with Lysotracker only.</figcaption>
</figure>
The cells imaged after the 4 hour incubation displayed a weaker Lysotracker signal than at t = 0. There were weak signals in the 640 channel as well. However, due to the low intensities and high backgrounds, those are most likely residual fluorescence from the dye rather than something extraneous.
• Branch
<figure>
<img src="http://openwetware.org/images/6/6e/Expfig5.png"height="420" width="624"/>
<figcaption>Figure 5: Stained cells incubated with stained branch structures.</figcaption>
</figure>
The fluorescent signal from the Lysotracker, while not as strong as at t = 0, was still within ideal intensity. There were strong signals in the 640 channel as well, and the signals originated within viable cells. There were multiple points of co-localization between the 488 and 640 channels.
• Block O
<figure>
<img src="http://openwetware.org/images/9/9a/Expfig6.png"height="423" width="622"/>
<figcaption>Figure 6: Stained cells with stained Block O structures.</figcaption>
</figure>
<figure>
<img src="http://openwetware.org/images/7/7e/Expfig7.png"height="424" width="624"/>
<figcaption>Figure 7: Stained cells with Block O structures showing structures that were not yet uptaken.</figcaption>
</figure>
The images for Block O at t = 4 shared many similar characteristics to those of for the branch, including multiple instances of co-localization between the two channels. However, in certain cases, a strong fluorescent signal was observed in the 640 channel located at the very edge of the cell membrane. This was not observed in any samples containing the branch structure.
Discussion:
In previous versions of the experiment, high cell concentration has always presented a problem. After attempting the experiment with 250,000 and 100,000 cells, 75,000 cells were used for this experiment. As a result, the cell concentration was ideal and single cells could be analyzed without interference from debris. However, the distribution of cells was not consistent, as there were spots of heavy density and spots where no cells were present. Cell viability was also better than previous experiments, possibly due to the lower cell concentration, and possibly due to an ideal concentration of FBS.
However, in cells where uptake was observed, the cell morphology was less than ideal. Cells displayed a non-circular membrane and granular cytoplasm, signs of an ailing cell. Since structures used did not contain miR-21 complementary overhangs, this could not have been due to the sequestration of miRs. Therefore, either the act of uptake caused the cells to lose ideal morphology, or structures are preferentially uptaken in ailing cells.
Many instances of dye colocalization, and therefore uptake, were observed in both the Block O and Branch treatments. However, more examples of uptake were observed in the Branch treatment. In addition, in the Block O treatment, there were instances where, after 4 hours of incubation, there were structures visible lining the membrane, perhaps in the process of being uptaken. While this is preliminary, the combination of fewer concrete instances of uptake and presence of structures still attempting to be uptaken hints that the Block O structures may not be as preferentially uptaken as the Branch structures, or may be uptaken at a slower rate. Quantitative analysis of uptake must be conducted to draw any conclusions on that matter.
For future experiments, overnight time lapse experiments may provide further insight on the mechanism and rate of uptake. The cells could also be nucleofected with fluorescent miR-21 to highlight the mechanism of miR sequestration and combine both structure uptake and cellular uptake in one experiment<A NAME="scroll2"></A>.
2. miR-21 Sequestration Experiment
Treatments
Branch
<figure>
<img src="http://openwetware.org/images/2/25/2.1.png"height="52" width="460"/>
</figure>
Procedure
• Calculate fluorescent miR-21 volume required to obtain the concentration ratios used for the treatment. Add miR-21 to 6 uL of structure and bring up volume to 18 uL with dH¬¬2O.
o Structure concentration: 1 nM
o miR-21 concentration: 100 nM
o miR-21 volumes;
<figure>
<img src="http://openwetware.org/images/4/4d/2.2.png"height="88" width="462"/>
</figure>
• Combine structures, miR-21, and dH2O and incubate at 37 C for 30 min while shaking.
• Prepare an agarose gel without EtBr according to protocol.
• After incubation, load 18 uL of samples into gel and run at 70 V for 2 hours.
o As controls, load 1 kb.p. DNA ladder, 7249 scaffold (branch), 8064 scaffold (Block O), and fluorescent miR-21 only.
• Remove gel from apparatus and image using a laser scanner capable of fluorescence detection.
• After fluorescent imaging, post-stain gel using SYBR Gold solution for 40 minutes and image.
Results
<figure>
<img src="http://openwetware.org/images/b/bc/2.3.png"height="258" width="484"/>
<figcaption>Figure 1: agarose gel image taken using bulk fluorescence.</figcaption>
</figure>
The miR-21 used for this experiment were tagged with the Atto 488 fluorescent dye. The image was taken using the Typhoon FLA 7000 gel imager.
<figure>
<img src="http://openwetware.org/images/3/31/2.4.png"height="90" width="422"/> <figcaption>Figure 2: agarose gel image taken after staining with SYBR gold.</figcaption> </figure>
The gel was stained in 10,000x SYBR Gold bath for 40 minutes before imaging.
Discussion
This experiment was designed to test the hypothesis that the complementary overhangs on the structures would successfully be able to bind to miR-21. It was also hypothesized that the structures with scrambled overhangs would not be able to sequester any miR-21 from solution. As figure 1 shows, there was no detectable fluorescence in the structure band in the lane with miR-21 and structures with scrambled overhangs, thus reinforcing the idea that the complementary overhangs are what sequesters the miR from solution.
By comparing figures 1 and 2, it is possible to conclude that, in samples containing both structures and fluorescent miR-21, the miR-21 fluorescence is colocalized with the structure bands in the gel. This shows that the structures were indeed able to remove the miR-21 from the solution.
In addition, the images also allow for conclusions to be drawn about the efficiency of sequestration. Figure 1 shows very little fluorescence beyond the structure band in the lanes with 0.33x, 0.5x and 1x samples. However, from the 2x lane onwards, there is a trend of increasing fluorescent signal beyond the structure band, suggesting that excess miR-21 is moving further along the gel. Since the increase in excess miR began after the 1x concentration, it can be concluded that the structure was able to sequester quantitative amounts of miR from the solution.
In the future, the miR sequestration experiment should be conducted with the Block O structures as well as the branch. While the experiment shows the ability of branch structures in aqueous solutions, future experiments should test for sequestration in solutions analogous to the interior of the cell, such as in cell lysate. Another avenue to test both cellular uptake and miR-21 uptake would be to nucleofect cells with fluorescent miR-21, incubate the cells with fluorescent structures, and then image them using fluorescence microscopy<A NAME="scroll3"></A>.
3. Cell Viability Experiment
Treatments:
● Cells only: Cells incubated in media and TAE buffer.
● Branch: Cells incubated in media with branch scrambled and branch miR structures in TAE buffer.
● Block-O: Cells incubated in media with block-o scrambled and bloc-o miR structures in TAE buffer.
Procedure:
● Cell Preparation:
○ Count cells using hemocytometer and trypan blue dye
○ Separate 2*105 cells for each sample
○ Wash cells twice with PBS and resuspend in warm media
● Structure Preparation:
○ Dilute structures to desired concentration in TAE with MgCl2
● Structure Addition:
○ Add cells in media to plate for incubation
○ Add structures to wells
○ Incubate for desired time
● Staining:
○ Add poly-L-lysine to wells in imaging plate and allow it to sit for 15 minutes
○ Remove poly-L-lysine and wash wells with PBS
○ Transfer cells from incubation plate to imaging plate
○ Add 1.75uL of Propidium Iodide (PI) to each sample for every 100 uL of solution
○ Allow the samples to sit, covered from light, for 15 minutes
○ Image the samples using the 488 nm laser
Hypothesis: Cells exposed to miR-complement structures should show decreased viability because of an increase in apoptosis-inducing proteins. Cells exposed to scrambled structures should show no change in viability when compared to a cells only control.
Results:
All images were taken after 24 hours of incubation at .5 nM final structure concentration. Structures were suspended in 50 uL of TAE buffer and added to cells. 50 uL of pure TAE was added to the cells only control to control for the addition of solution.
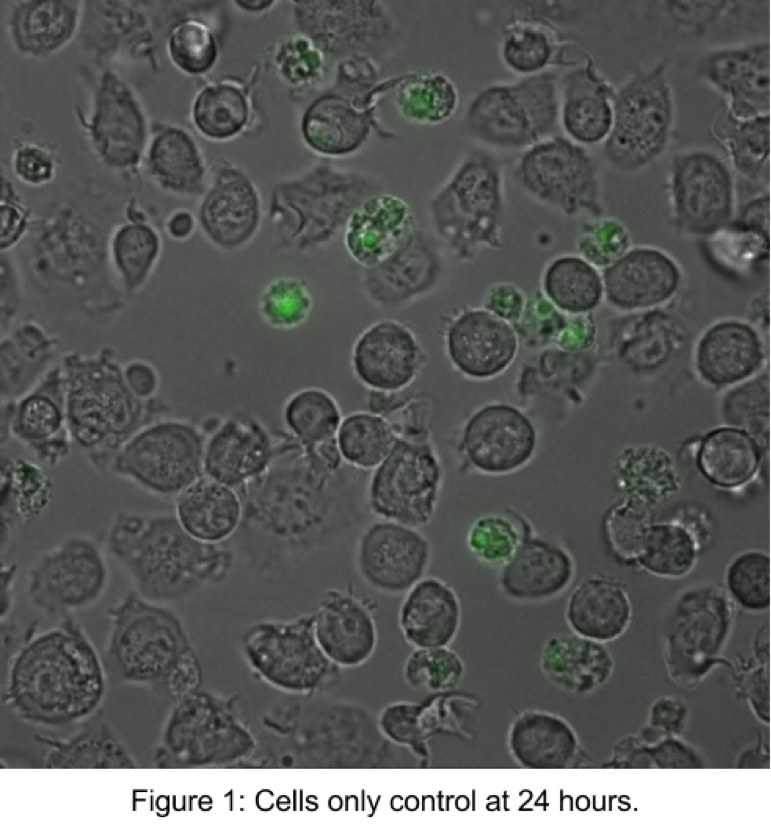
● Cells only:
<figure>
<img src="http://openwetware.org/images/7/7b/Cv1.png"height="422" width="422"/> </figure>
The cells only control showed viability of around 85%. Shown above (Figure 1) is an example of a composite image of DIC and 488 nm. PI excited at 488 nm (red) and emits at 561 (green), so dead cells are highlighted in green.
● Branch with miR-21 complement overhangs:
<figure>
<img src="http://openwetware.org/images/a/ab/Cv2.png"height="422" width="422"/> </figure>
The cells showed about 50% viability when treated with .5 nM branch structures with miR complement overhangs, a marked decrease in viability. Overall, there is much more fluorescence and cell morphology supports the decreased viability.
● Branch with scrambled overhangs:
<figure>
<img src="http://openwetware.org/images/4/4b/Cv3.png"height="422" width="422"/> </figure>
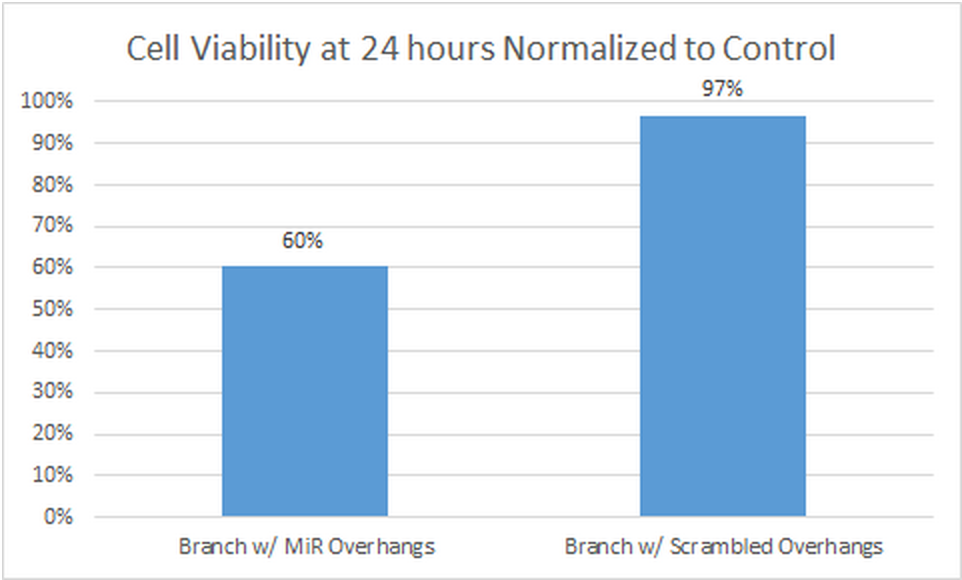
These cells show viability around 80%, close to that exhibited by the cells only control. Their morphology overall supports the lower fluorescence when compared to the miR complement sample. A comparison of viability data normalized to the cells only control.
<img src="http://openwetware.org/images/1/10/3.1.png"height="280" width="462"/>
<figcaption>Comparison of cell viability after 24 hour incubation normalized to cells only control</figcaption>
</figure>
Discussion:
Previously, higher quantities of cells had been studied for viability. However, these trials resulted in crowded images that were impossible to analyze because so many cells were in different focal planes. Low cell viability was also an issue in all samples. 2*105 cells were used because that resulted in reasonable overall viability and clear images. Propidium iodide staining also had to be optimized. Low concentrations yielded very little signal, while overstaining resulted in saturated images that gave little useful information.
The images shown above are representative of the images in general. Through counting, marked decreases in viability were observed in the cells treated with the miR complement branch structure at 24 hours. Other qualitative data supports this conclusion. The morphology of the miR complement cells showed that the cells were generally unhealthy, showing irregular shape and granularity of the cytoplasm. These characteristics were not nearly as common in the under the other conditions. The histogram for signal intensity of each set of images also suggests that there was a much higher signal in the miR complement samples. The 488 signal peaked at a higher intensity and continued to higher intensities in the miR samples, while the other samples showed lower peaks and no signal at higher intensities.
Future experiments should include more time points, primarily, along with varying structure concentrations. Higher structure concentrations should yield a greater decrease in cell viability. Data at other timepoints would be useful if correlated with data on protein expression and uptake. It would also be useful to use flow cytometry to count cells. Flow cytometry measures individual cells, known as “events”, that pass by a detector. It can measure morphology and fluorescence to get a better measurement of cell viability and is the accepted technique for measuring cell viability in literature<A NAME="scroll4"></A>.
4. PTEN Protein Expression
Treatments:
<figure>
<img src="http://openwetware.org/images/f/fe/1_treattable.png"height="174" width="434"/>
</figure>
Procedure:
• Cell preparation and incubation:
o Count cells and aliquot ~1 million cells per sample.
o Wash cells twice and resuspend in 900 uL of cell media.
o For cells only samples, add 100 uL of TAE buffer. For other samples, add 100 uL of 1 nM structures to the corresponding sample.
o Plate samples in 24 well plate and incubate at 37 C for the desired timepoint (24, 48 or 72 hrs).
• Protein extraction
o After incubation, wash cells twice in PBS, and resuspend in 100 uL Protein Lysis buffer. Mix vigorously by pipetting.
• Lysis buffer preparation: add PMSF in a 1:100 dilution, and protease inhibitor in a 1:200 dilution into lysis buffer.
o Incubate cells in Lysis buffer on ice for 10 min.
o After incubation, centrifuge sample at 10,000 g for 15 mins.
• Protein quantification using BCA assay.
o Prepare BCA assay solution for all samples in a falcon tube
• 200 uL reagent A for each sample, 1:50 dilution of reagent B.
o Prepare standards by serially diluting bovine serum albumin to known concentrations.
o In a 96 imaging plate, add 200 uL of BCA assay solution and 10 uL of sample or standard.
o Incubate at 37 C for 30 min.
o Image plate using a plate reader.
• Protein separation using Polyacrylamide gel electrophoresis.
o Create a 10% Polyacrylamide gel according to protocol.
o Mix 6.5 ug protein from each sample with 10 uL Laemmli solution, and place on heating block at 95 C for 5 mins.
o Prepare biotinylated ladder by mixing 1 uL of ladder solution with 15 uL of Laemmli solution.
o On gel, load 10 uL kaleidoscope ladder, 11 uL biotinylated ladder, and the entire volume for the samples. Run at 60 V until sample enters gel front. Then, run at 75 V for 2 hours.
• Protein transfer to membrane.
o Make transfer buffer by adding 100 mL 10x transfer buffer, 200 mL methanol, and 700 mL dH2O.
o Cut out membrane of proper size.
o Charge membrane by washing in methanol for 15 s, then water for 5 min, then soak in transfer buffer.
o Assemble transfer apparatus in correct order. From top to bottom: foam pad, Whatman paper, membrane, gel, Whatman paper, foam pad.
• When placing gel, ensure that gel is not dry for too long, and that it lines up with membrane.
o Place assembled apparatus into transfer buffer and run in ice at 100 V for 1 hour.
• Incubation with primary antibodies (PTEN).
o Remove membrane from transfer apparatus.
o Wash once with water, and 4-5 times with TBST buffer for 5 min each on a rocker.
o Prepare a 1:1000 dilution of PTEN antibody in 5% milk as blocking agent.
o After washes, add 10 mL of antibody solution to membrane.
o Incubate membranes in antibody solution on rocker at 4 C overnight.
• Incubation with secondary antibodies (PTEN).
o Remove primary antibody solution from membranes.
o Wash 4-5 times with TBST buffer on rocker with 10 min for each wash.
o Prepare 1:1000 dilution of secondary anti-rabbit antibodies and 1:2000 dilution of anti-biotin antibodies in 1% milk as blocking agent.
o After washes, add 10 mL of secondary antibody solution to membranes.
o Incubate on rocker at room temperature for 1 hour.
• Imaging (PTEN)
o Remove secondary antibody solution from membranes.
o Wash 5-10 times with TBST buffer on rocker.
o Prepare developing solution.
• 4.5 mL ddH2O, 250 uL developing buffer 1, and 250 uL developing buffer 2.
o After wash, blot membrane to remove excess liquid and lay membrane on flat surface.
o Add developing solution on membrane until uniformly covered.
o Leave developing solution on membrane for 20 s, then blot off excess liquid.
o Lay membrane on cassette, wrap in plastic wrap, and close cassette to protect from light.
o In dark room, place film on top of membrane inside cassette, and let film develop for ~30 s.
o Remove film, and place in developer to obtain image.
• GAPDH Detection
o Follow same procedure for primary antibody incubation and secondary antibody incubation using the anti-GAPDH antibody instead of the anti-PTEN antibody. For imaging, develop film ~3 s instead ~30 s.
Results:
• Total protein concentrations using BCA protein assay.
<figure>
<img src="http://openwetware.org/images/3/31/2bcacurve.png"height="306" width="446"/>
<figcaption>Figure 1: BCA standard curve using bovine serum albumin dilutions of known concentrations.</figcaption>
</figure>
The linear trendline for the standard curve fit well with the experimental data, with a coefficient of correlation over 0.99. However, the resolution of the data did not extend to the lowest concentration tested (0.031 ug/uL).
<figure>
<figcaption>Table 1: Total protein concentrations in ug/uL in different samples calculated using the BCA protein assay.</figcaption>
<img src="http://openwetware.org/images/a/ad/3_table1.png"height="160" width="572"/>
</figure>
The total protein concentrations for the different samples showed high, random variability.
• Western blot analysis
24 hrs
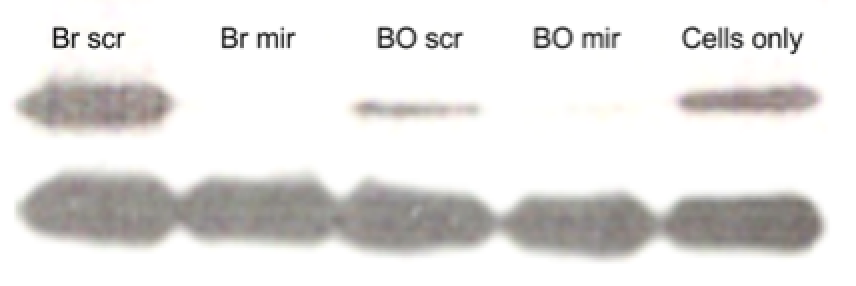
<figure>
<img src="http://openwetware.org/images/4/4e/4fig2.png"height="134" width="438"/>
<figcaption>Figure 2: Western blot images showing relative expression of PTEN and GAPDH. The top bands are PTEN, and the bottom bands are GAPDH.</figcaption>
</figure>
The images of the blots were ideal, with specific antibody binding, resulting in clear bands and low background. The 5% milk blocking solution proved to be better than the bovine serum albumin solution, which caused non-specific binding of antibodies, resulting in high levels of uneven background levels.
For the PTEN bands, the antibody stained well, with sharp intense bands. The band front was slightly crooked, but was probably the result of loading errors rather than differences in the protein composition. Qualitatively, the Br mir sample displayed the highest concentration of PTEN levels, whereas the cells only sample showed the least. For BO, the scr sample seemed to have higher intensity, suggesting a higher expression of PTEN relative to the mir sample.
For the GAPDH bands, the antibody staining was not ideal, since the bands were not sharp and showed variable intensity. The development time was also too high, since the bands bled into each other. In terms of expression, the different samples seemed to show similar levels of band intensity, suggesting that the treatment did not affect GAPDH expression
<figure>
<figcaption>Table 2: Densitometric analysis of western blot band intensities to determine PTEN expression relative to housekeeping protein (GAPDH) and different controls.</figcaption>
<img src="http://openwetware.org/images/a/a2/5table2.png"height="176" width="458"/>
</figure>
<figure>
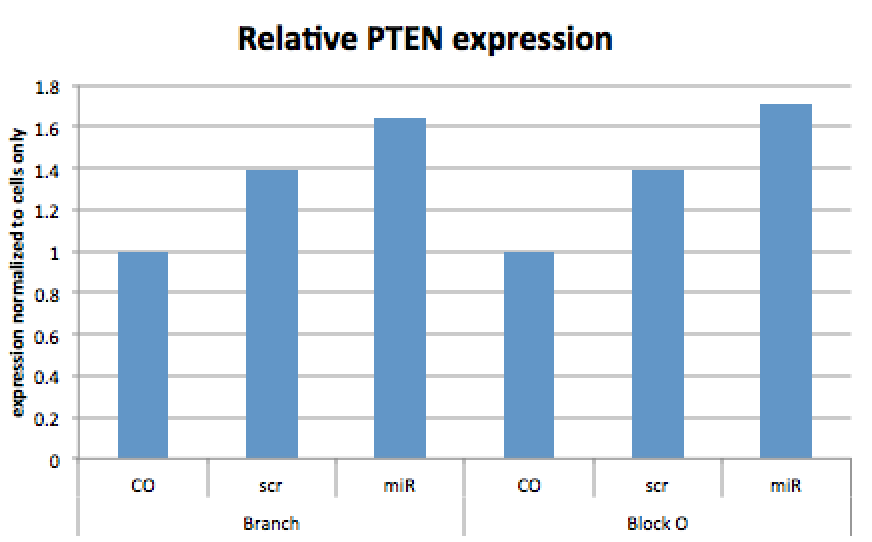
<img src="http://openwetware.org/images/c/cb/6fig3.png"height="348" width="496"/>
<figcaption>Figure 3: Graph showing relative expressions of PTEN in cells incubated with different structures after 24 hours incubation.</figcaption>
</figure>
<figure>
<img src="http://openwetware.org/images/0/0d/7fig4.png"height="346" width="494"/>
<figcaption>Figure 4: Graph showing relative expressions of PTEN in cells incubated with different structures after 24 hours incubation.</figcaption>
</figure>
The densitometric analysis confirmed the qualitative assessments made using the blot images. After normalization to the GAPDH band, the highest intensity band was the Br scr, with the lowest intensity being the cells only sample (Table 2). Further normalization to the cells only control showed that all four structure treatments caused a significant increase in the PTEN levels, as shown in Figure 3. However, further normalization to the scrambled overhangs control highlighted the discrepancies between the scrambled and the complementary mir treatments for both structures. For the branch structures, the mir treatment showed an increase in PTEN relative to the scrambled treatment, whereas for Block O the mir treatment showed lower levels of PTEN expression relative to its corresponding scrambled treatment as seen in Figure 4.
48 hrs
<figure>
<img src="http://openwetware.org/images/3/33/8fig5.png"height="134" width="438"/>
<figcaption>Figure 5: Western blot images showing relative expression of PTEN and GAPDH. The top bands are PTEN, and the bottom bands are GAPDH.</figcaption>
</figure>
The characteristics of the blots were at 48 hours were very similar to those at 24 hours, with one significant difference. The sample with the most band intensity was the BO mir sample instead of the Br mir sample. In addition, the BO mir band was visibly more intense than the BO scr sample, while the there was no discernible differences between the intensities of the Br scr and Br mir bands. However, the GAPDH band for the Br scr sample seemed more intense than the that of the Br mir band.
<figure>
<figcaption>Table 3: Densitometric analysis of western blot band intensities to determine PTEN expression relative to housekeeping protein (GAPDH) and different controls.</figcaption>
<img src="http://openwetware.org/images/d/d3/9table3.png"height="150" width="458"/>
</figure>
<figure>
<img src="http://openwetware.org/images/0/08/10fig6.png"height="342" width="472"/>
<figcaption>Figure 6: Graph showing relative expressions of PTEN in cells incubated with different structures after 48 hours incubation.</figcaption>
</figure>
<figure>
<img src="http://openwetware.org/images/4/48/11fig7.png"height="344" width="494"/>
<figcaption>Figure 7: Graph showing relative expressions of PTEN in cells incubated with different structures after 24 hours incubation.</figcaption>
</figure>
Even though no qualitative conclusion could be drawn about the Branch structures from the blot images, densitometry revealed that the Br mir sample, when normalized to GAPDH, did indeed show a higher PTEN expression relative to the scrambled overhangs expression. Furthermore, in contrast to the 24 hr time point, the BO mir sample also showed a higher level of expression than the BO scr sample. In fact, the increase in expression in the BO sample was greater than that of the Br sample (Figure 7). One continuing trend from the 24 hr time point was the higher level of PTEN expression compared to the cells only sample (Figure 6).
72 hrs:
<figure>
<img src="http://openwetware.org/images/9/9e/12fig8.png"height="134" width="438"/>
<figcaption>Figure 8: Western blot images showing relative expression of PTEN and GAPDH. The top bands are PTEN, and the bottom bands are GAPDH.</figcaption>
</figure>
At 72 hours, the blot images were significantly different than the other two time points. Once again, the blots were clear with no background. However, in terms of band intensity, the Br scr PTEN band was significantly more intense than all others, where PTEN bands for Br and BO mir samples were non-existent, suggesting that there was no PTEN present in those cells. The GAPDH bands looked similar to those from the 24 and 48 hr time points.
Discussion:
The BCA protein assay is conducted to measure the concentrations of total protein in unknown protein extract samples so that equal amount of protein can be used for western blot experiments, thus reducing the number of confounding factors. The assay calculates concentration by comparing the absorbance from the unknown sample to a standard curve generated by samples with known concentrations. The resolution of the assay is therefore equal to the range on the standard curve. The standard curve generated for this experiment had an effective range between 0.125 ug/uL and 2 ug/uL, since the 0.031 ug/uL sample did not fit with the linear trend. However, this was not an issue, since all the unknowns measured had concentrations above 0.125 ug/uL.
The branch and Block O structures with miR-21 complementary overhangs were developed with the intention of sequestering free miR-21 from the cell cytoplasm. Since one of most well studied effects of miR-21 over-expression is the suppression of PTEN protein, it was hypothesized that the level of PTEN expression would increase in samples treated with the mir structures compared to controls such as structures with scrambled overhangs or cells incubated without any structures.
At 24 hours, there was a measurable increase in relative PTEN levels in the branch mir treatment compared to the scr or the cells only treatment, suggesting that the branch treatment was successful in suppressing miR-21 and increasing the levels of PTEN protein expression. At 48 hours, both Branch and Block O mir treatments showed an increase in relative PTEN expressions, further reinforcing the positive outlook for the structures.
However, a few issues persisted. At 24 hours, the PTEN expression in the BO scr control was higher than that of the BO mir treatment, suggesting that the mir structure was unsuccessful in sequestering miR-21 at 24 hours, even though they were successful in doing so at 48 hours. While this may seem contradictory, cellular uptake experiments conducted earlier hinted that the Block O structures were possibly uptaken by cells at a slower rate than the branch structures. If so, it is possible that the Block O structures were not present in cells at the threshold levels necessary to cause a significant decrease in miR-21 at 24 hours, but were present at required levels 48 hours after incubation. In addition, even though the BO mir treatment was unsuccessful at 24 hours, at 48 hours it displayed a higher increase in PTEN than the branch structure. This can possibly be explained by the fact that the Block O structure has more overhangs than the branch structure. If, at 48 hours, the concentrations of Block O and Branch had equilibrated to a similar number, the 12 extra complementary overhangs on the Block O structure may explain the greater effect seen in the Block O sample compared to the branch sample.
Another discrepancy was the increased levels of PTEN in the scrambled control relative to the cells only control. Ideally, neither the structures nor the scrambled overhangs on the structures should have any extraneous effect on the cell, so the cells only and the scrambled overhangs controls should effectively be the same. However, since an increase in PTEN was observed, the structures themselves could be having an unexpected additive effect to the regulation of PTEN. To test this theory, further replicates of the present experiment and experiments with structures without any overhangs must be conducted in the future.
Finally, the result for the 72 hour time point study was unexpected, since the PTEN levels in the mir treatment were completely suppressed, whereas the PTEN levels, especially in the Br scr treatment, were highly elevated. Initially, it was though that this could have been caused by a significant loss of viability in the mir treatments due to the increase in PTEN. However, such an event would also have reduced the amount of housekeeping protein (GAPDH) in the sample, which was not observed. Another possibility may be that the apoptotic mechanism itself reduces the amount of PTEN in cells, and thus shows decreased expression levels at extended time points such as 72 hours. However, further replicated of this experiment and further research into literature must be conducted to draw a conclusion about the causes behind such a phenomenon.
This experiment represents only one replicate, and is therefore not statistically significant. To achieve statistical significance, comparable results must be obtained in at least two or more replicates of the same experiment<A NAME="scroll5"></A>.
5. PTEN mRNA Experiment
Treatments:
<figure>
<img src="http://openwetware.org/images/9/9d/1-table.png"height="134" width="438"/>
</figcaption>
</figure>
Procedure
• Structure incubation
o Count cells and aliquot ~1 million cells per sample.
o Wash cells twice using PBS.
o Resuspend cells in 900 uL of cell media.
o Add 100 uL of structures at 1 nM to cells.
o Incubate for desired amount of time at 37 C.
• RNA isolation
o Wash cells twice using PBS.
o Resuspend cells in 1 mL Trizol reagent, vortex thoroughly for 1 minute, and incubate at room temperature for 5 minutes.
o Add 200 uL chloroform in each sample, vortex briefly to mix, incubate at room temperature for 3 minutes, and centrifuge at 12,000 g for 10 minutes.
o Pipet out the clear aqueous phase, being careful not to suck up any of the buffy coat or Trizol.
o Add 500 uL of 100% isopropanol and 2.5 uL glycogen (20 mg/mL) into the tube, incubate at room temperature for 10 minutes, and centrifuge at 12,000 g for 10 minutes.
o Aspirate supernatant and resuspend in 1 mL 75% ethanol. Centifuge at 7500 g for 5 minutes.
o Aspirate out supernatant and dry pellet until ethanol has evaporated. Resupend pellet in distilled water.
o Measure the RNA concentration in each sample.
• DNAse Treatment
o Add 2.5 ug of extracted RNA into 2.5 uL of DNAse I, 2.5 uL of 10x DNAse buffer, and balance of water to get final volume of 25 uL.
o Incubate at room temperature for 15 minutes.
o Add 2 uL of 25 mM EDTA and heat at 65 C for 10 minutes to deactivate DNAse.
• Reverse Transcriptase(RT) PCR
o Create master mix using RT buffer, dNTP mix, random hexamers, RNAse inhibitor, and reverse transcriptase enzyme according to protocol.
o Add 14 uL of DNAse treated RNA sample into 6 uL of master mix.
o Incubate for 1 hour at 37 C.
• qPCR on cDNA
o Prepare qPCR master mix using qPCR buffer, dNTP mix, MgCl2 solution, forward and reverse primers, taq-polymerase enzyme and water according to protocol.
• 3 mM final concentration of Mg¬2+.
• 0.5 uM final concentration for primers.
o Add 2 uL of cDNA solution to 18 uL of master mix in a 96-well imaging plate. Mix by pipetting.
o Centrifuge plate at 2000 rpm for 15 s before placing in qPCR machine and starting reaction.
o For reaction with PTEN primers:
• Denaturing at 95 C for 10 s.
• Annealing at 52 C for 2 s.
• Extending at 70 C for 15 s.
o For reaction with Actin primers:
• Denaturing at 95 C for 10 s.
• Annealing at 60 C for 2 s.
• Extending at 70 C for 15 s.
• Standard curve generation
o Create serial dilution of known concentrations for both PTEN and Actin samples.
o Run qPCR reaction on standards with the same conditions as unknown samples, setting a threshold fluorescence level to detect.
o Plot the concentration of standards against the cycle number (Cq) needed to reach the threshold fluorescence for those standards. Perform a linear regression on the data.
Results
RNA concentration
<figure>
<figcaption>Table 1: Total RNA concentrations and absorbance ratio between 260 nm and 280 nm in samples after 12 hour incubation.</figcaption>
<img src="http://openwetware.org/images/b/bc/2table1.png"height="134" width="438"/>
</figure>
<figure>
<figcaption>Table 2: Total RNA concentrations and absorbance ratio between 260 nm and 280 nm in samples after 24 hour incubation.</figcaption>
<img src="http://openwetware.org/images/8/88/3table2.png"height="134" width="438"/>
</figure>
For samples lysed after the 12 hour incubation, the total RNA was resuspended in 40 uL ddH2O. For samples lysed after the 24 hour incubation, the total RNA was resuspended in 20 uL ddH2O, leading to a larger concentration. In addition, during extraction of the aqueous phase, the total only 400 uL of the total aqueous phase was removed from the 12 hr incubation samples, whereas almost the entire aqueous phase was extracted from the 24 hr incubation samples.
Actin Melt Curve
• 12 hrs
<figure>
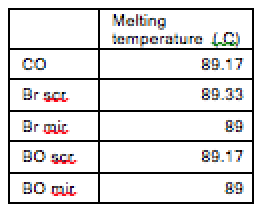
<figcaption>Table 3: Actin gene melting temperatures for different samples.</figcaption>
<img src="http://openwetware.org/images/2/2d/4table3.png"height="112" width="120"/>
</figure>
<figure>
<img src="http://openwetware.org/images/4/4b/5fig1.png"height="224" width="338"/>
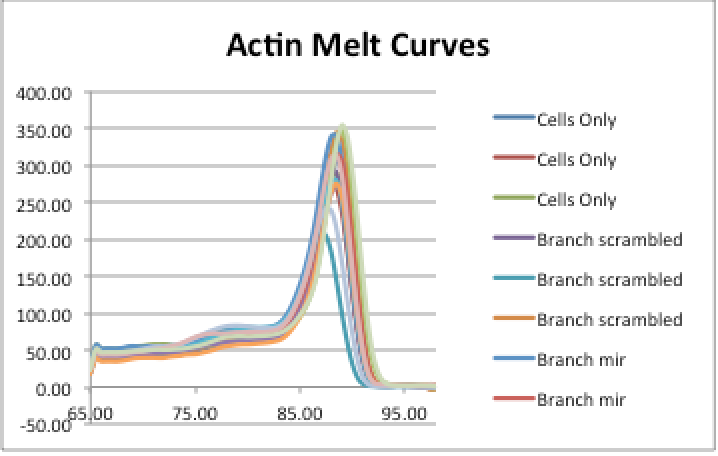
<figcaption>Figure 1: Melt curves for the Actin amplicon in the different samples. Includes replicates.</figcaption>
</figure>
• 24 hrs
<figure>
<figcaption>Table 4: Melting temperatures of the Actin amplicon.</figcaption>
<img src="http://openwetware.org/images/8/8a/6table4.png"height="100" width="120"/>
</figure>
The melting temperatures at t = 24 hrs show a higher variability than those at t = 12 hrs. The values are also consistently lower than their counterparts at t = 12 hrs.
<figure>
<img src="http://openwetware.org/images/6/6b/7fig2.png"height="220" width="300"/>
<figcaption>Figure 2: Melt curve for the Actin amplicon for different samples. Includes replicates.</figcaption>
</figure>
Actin Quantification Curves
• 12 hrs
<figure>
<img src="http://openwetware.org/images/d/d4/8fig3.png"height="200" width="320"/>
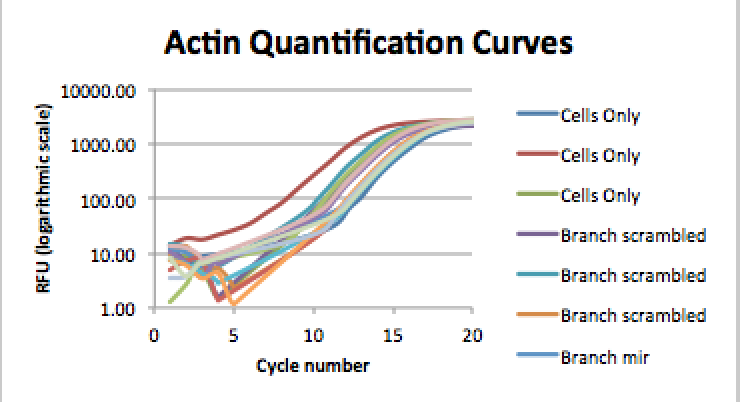
<figcaption>Figure 3: Quantification curve for the Actin amplicon. Replicates included.</figcaption>
</figure>
The amplification curves show that the sample with the highest absolute levels of PTEN was the Cells only sample. Overall, there was no notable difference in the cycle numbers among the different samples, and so there was no notable difference in concentration either. There was, however, a relatively large discrepancy in concentration among replicates within the sample.
• 24 hrs
<figure>
<img src="http://openwetware.org/images/e/e6/9fig4.png"height="222" width="350"/>
<figcaption>Figure 4: Quantificaion curve for Actin amplicon. Replicates included.</figcaption>
</figure>
As with the quantification curves for the 12 hr experiment, the different replicates did not share the same concentrations. In addition, the linear section of some of the curves were not smooth, indicating the possible presence of contaminants in those samples.
Actin Standard Curve
<figure>
<img src="http://openwetware.org/images/d/d6/10fig5.png"height="210" width="350"/>
<figcaption>Figure 5: Standard curve for Actin amplicon.</figcaption>
</figure>
The Actin standard curve shows a high coefficient of correlation at 0.98, meaning that the standards prepared were of the right concentration. However, the efficiency of the qPCR reaction, which is a function of the slope, is 154% which is well outside the acceptable range. This suggests that the conditions for the reaction are not ideal, and may also hint at primer dimer formations.
PTEN Melt Curves
• 12 hrs
<figure>
<figcaption>Table 5: Average melting temperatures for PTEN amplicon.</figcaption>
<img src="http://openwetware.org/images/a/a3/11table5.png"height="100" width="150"/>
</figure>
<figure>
<img src="http://openwetware.org/images/f/f5/12fig6.png"height="200" width="438"/>
<figcaption>Figure 6: Melt curve analysis for PTEN amplicon at 12 hours. Replicates included.</figcaption>
</figure>
• 24 hrs
<figure>
<img src="http://openwetware.org/images/7/76/13fig7.png"height="250" width="338"/>
<figcaption>Figure 7: Melt curve analysis for PTEN amplicon at 24 hours. Replicates included.</figcaption>
</figure>
As the curves show, the purity of the 24 hr samples were inferior to those at 12 hrs. The melting temperatures were shifted to the left, the peaks were broader than ideal, and some samples had multiple peaks, indicating the presence of a second amplification product. To promote validity of data, samples that showed multiple melting temperatures were excluded from further analysis.
PTEN Quantification Curves
• 12 hrs
<figure>
<img src="http://openwetware.org/images/f/fb/14fig8.png"height="200" width="438"/>
<figcaption>Figure 8: Quantification curves for Actin amplicon at 12 hours. Replicates included.</figcaption>
</figure>
• 12 hrs
<figure>
<img src="http://openwetware.org/images/e/ef/15fig9.png"height="250" width="438"/>
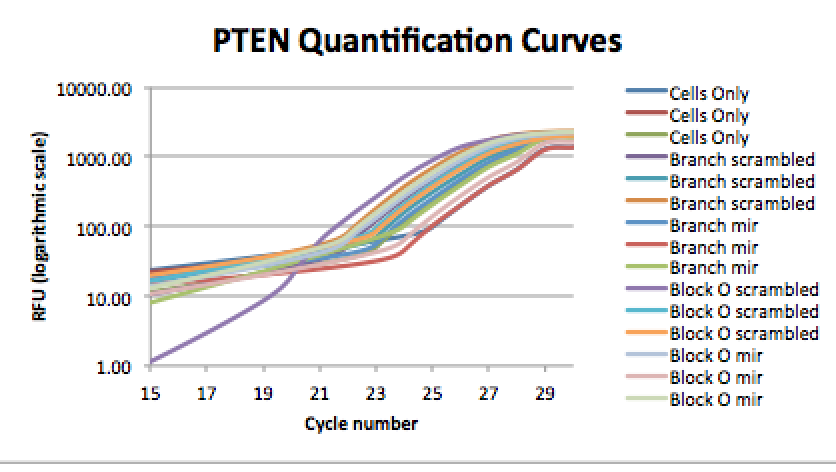
<figcaption>Figure 9: Quantification data for PTEN amplicon at 24 hrous. Replicates included.</figcaption>
</figure>
PTEN Standard Curve
<figure>
<img src="http://openwetware.org/images/e/ea/16fig10.png"height="250" width="438"/>
<figcaption>Figure 10: Standard Curve for PTEN amplicon.</figcaption>
</figure>
The PTEN standard curve also has a high correlation of coefficient. In addition, the efficiency of the qPCR reaction is at 105% which is within the accepted range of efficiencies. This means that the data obtained from the PCR reaction is reliable.
Analysis
• 12 hrs
<figure>
<figcaption>Table 6: Comparative analysis of PTEN mRNA expression relative to Actin mRNA expression in different treatments.</figcaption>
<img src="http://openwetware.org/images/d/d2/17table6.png"height="114" width="338"/>
</figure>
<figure>
<img src="http://openwetware.org/images/3/3b/18fig11.png"height="300" width="338"/>
<figcaption>Figure 11: Bar graph showing relative PTEN mRNA expression normalized to the expression of cells only sample.</figcaption>
</figure>
• 24 hrs
<figure>
<figcaption>Table 7: Comparative analysis of PTEN mRNA expression relative to Actin in different treatments.</figcaption>
<img src="http://openwetware.org/images/c/c3/19table7.png"height="134" width="438"/>
</figure>
<figure>
<img src="http://openwetware.org/images/d/d9/20fig12.png"height="300" width="438"/>
<figcaption>Figure 12: Bar graph showing relative PTEN mRNA expression normalized to the expression of cells only sample.</figcaption>
</figure>
Discussion
The RNA isolation and DNAse I treatment of cell lysate is a crucial step in mRNA analysis using qPCR, since those steps are the main determinants of the purity of the sample being amplified. In particular, separating the RNA aqueous phase from the rest during isolation can be difficult, since it is usually discouraged to separate the top phase of suspensions first, and is very easy to accidentally remove some of the buffy coat or Trizol layer in the process. For the 12 hr data point, efforts were made to reduce the amount of contamination by refraining from removing the entire aqueous phase. For the 24 hour data point, efforts were made to remove as much of the aqueous phase as possible.
RNA quantification shows the consequence of the two procedures. The total RNA concentration for the 12 hr time point was consistently lower than that of the 24 hr time point, showing that a high yield was sacrificed in the attempt of reducing contamination. However, comparing the melt curves produced after amplifying the two sets of samples shows that the purity of the 12 hr data set was a lot higher than the 24 hr data set. Therefore, even though yield decreased, the purity of the samples obtained greatly increased. Since even with the low yield there was sufficient quantities of RNA for a triplicate experiment, it is advisable to aim for higher purity rather than higher yield.
Due to the lower purity of the samples, the melt curves and quantification curves for both the PTEN and Actin PCR were not ideal. However, while the PTEN data showed marked decrease in quality, the Actin data was relatively more resistant to the issues due to the lower purity of the sample. Since all other factors were consistent between the two amplifications, this trend suggests that the Actin primers may display a higher specificity than the PTEN primers, and are therefore able to function relatively well even in the presence of contaminants. This is further supported by the melting temperatures of the Actin forward and reverse primers, which are only 2 C apart, and thus the annealing temperature can be honed in more precisely for them. In contrast, the melting temperatures for the PTEN primers are 10 C apart, which means that no annealing temperature can be ideal for both primers.
The 12 hr data did not suffer from issues of purity, and so the data was used to generate standard curves for both Actin and PTEN. For PTEN, the standard curve was ideal, showing an inverse relationship between cycle number and starting concentration, a high R2 value, and an efficiency that is within ideal range. However, the efficiency for the Actin qPCR reaction was much higher than accepted values, showing that there were inhibitors to the PCR reaction present in the sample. These inhibitors could be ethanol from the RNA isolation step, or secondary metabolites that could be present in the sample. To reduce the effect of inhibitors in the future, extra care should be taken in preparing the standards to ensure all the ethanol has evaporated out and the sample is free from any extraneous substances.
The hypothesis for this experiment was that miR-21 sequestration by successfully uptaken structures will result in an increase in the mRNA levels of PTEN, a primary target of miR-21. The 24 hour time point was initially chosen as a standard period of incubation. However, when western blot analysis showed an increase in PTEN levels for cells incubated with branch structures, a 12 hour time point was also chosen as it was hypothesized that the increase in mRNA would occur prior to the resultant increase in protein. Actin was chosen as a housekeeping gene since the levels of Actin would not change due to the treatments. The Cq data from the qPCR was used to normalize the expression of PTEN mRNA in the different samples to the expression of Actin, so that any differences in expression would only be due to the treatment. The normalized PTEN values in the treatment were then compared to the cells only control, and the scrambled overhangs control.
Figure 11 shows that, at 12 hours after incubation, there is no significant difference in PTEN mRNA among the different samples, for branch or Block O. Previous experiments showed that structures were successfully uptaken by cells as early as 4 hours after incubation, so any change in mRNA levels should be observable at 12 hours.
Figure 12 shows a slightly greater discrepancy between PTEN mRNA levels between the treatment and scrambled control, with a 5% difference in the Branch structures and an 8% difference in the Block O structures. This may suggest that the mechanism of miR sequestration is slower in cells than anticipated, so any changes would take longer to be observable.
In the future, more time points between and beyond 12 and 24 hours must be analyzed to capture the specific moment of mRNA increase, if any such moment exists. Further replicates must also be conducted for the studies done already to ensure statistical significance.
<A HREF="#top"><img src="http://openwetware.org/images/4/46/Arrow_up.png" id="fixedbutton"height="40" width="40"></A>
<a href="http://openwetware.org/index.php?title=Biomod/2014/OhioMOD/experimentnotes&action=edit">Edit Experiment Notes</a>
</body>
</html>
<html>
<head> <style>
- Footer table {
width: 96.8%; max-width:2350px; bottom:0px; height:50px; /* Height of the footer */ font-weight:300; background-color: #2E2E2E; text-align:center; color: white; font-size:13px; border: 3px white solid;
}
- Footer {
clear: both; /*may be omitted*/ position: absolute; bottom: 12px; background-color:#fffff; width: 100%; height: 40px; /* or anything you like */ }
</style>
</head>
</html>

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}