Biomod/2013/Harvard/protocols
<html>
<head>
<link href='http://fonts.googleapis.com/css?family=Open+Sans' rel='stylesheet' type='text/css'>
</head>
<style>
body {
font-family: 'Open Sans', sans-serif; overflow-y: scroll;
}
.container {
background-color: #ffffff; margin-top:0px
} .OWWNBcpCurrentDateFilled { display: none; }
h1 {
font-size: 36px; line-height: 36px; padding-top: 5px; border-bottom-width: 0;
}
h3 {
font-size: 18px;
}
h5
{
font-family: 'Open Sans', sans-serif; font-size: 11px; font-style: normal; text-align: center; margin:0px; padding:0px;
}
- column-content
{
/* Uncomment to Dewikify width: 0px; float: left; */ margin: 0 0 0 0; padding: 0;
} .firstHeading {
display:none; width:0px;
}
- column-one
{
display:none; width:0px; padding-top: 35px; background-color: #ffffff;
}
- globalWrapper
{
width: 900px; background-color: #ffffff; margin-left: auto; margin-right: auto
}
- content
{
margin-left: 0px; margin-top: 0px; padding-top: 0px; align: center; /*padding: 12px 12px 12px 12px; width: 30%; background-color: #ffffff; border: 0; */
}
- bodyContent
{
width: 850px; align: center; background-color: #fffffff;
}
- column-content
{
width: 900px; background-color: #ffffff;
}
- footer
{
position: center; width: 900px;
} @media screen {
body { background: #000000 0 0 no-repeat; /* changed default background */ }
}
- menu
{
align: left; width: 10em; padding: 0px 10px 10px 10px; background-color: #FFFFFF; float: left;
}
- pagecontent
{
width: 620px; min-height: 400px; float: left; margin-left: 0px;
}
.group:after {
content: ""; display: table; clear: both;
}
.editsection {
/*display: none*/
}
a:link {color:#FF6060;}
a:active {color:#B24343; }
a:hover {color:#B24343; text-decoration: none}
a:visited {color:#FF6060;} /* visited link */
/*Expanding list*/
- exp { list-style: none; }
- exp li {
height: 1.8em; border-top: 1px solid #dedede; margin: 0 0 0 0; padding-top: .2em
}
- exp li:hover { background-color: #F8F8F8}
- exp li a:hover { display: block }
</style> </html>
HARVARD
BIODESIGN
BIOMOD 2013
-
<html><a href="http://openwetware.org/wiki/Biomod/2013/Harvard">
- HOME </a></html> <html><a href="http://openwetware.org/wiki/Biomod/2013/Harvard/introduction">
- INTRODUCTION </a></html> <html><a href="http://openwetware.org/wiki/Biomod/2013/Harvard/design">
- DESIGN </a></html> <html><a href="http://openwetware.org/wiki/Biomod/2013/Harvard/methods">
- METHODS </a></html> <html><a href="http://openwetware.org/wiki/Biomod/2013/Harvard/protocols">
- PROTOCOLS </a></html> <html><a href="http://openwetware.org/wiki/Biomod/2013/Harvard/team">
- TEAM </a></html> <html><a href="http://openwetware.org/wiki/Biomod/2013/Harvard/lab">
- LAB NOTEBOOK </a></html> <html><a href="http://openwetware.org/wiki/Biomod/2013/Harvard/results">
- RESULTS </a></html> <html><a href="http://openwetware.org/wiki/Biomod/2013/Harvard/references">
- REFERENCES </a></html>
![]()

Protocols
Cell Culture Protocols
Bacterial Cell Cloning / Inoculation of Cells
Entire procedure is done under flame
- For small test tubes add:
- 5mL LB media
- 500µL 20% glucose (final conc. 0.2%)
- 7.35µL of CM (final conc. 50µg/mL, working stock: 34 mg/mL)
- Add the cells
- If from glycerol stock, stab with pipette tip.
- If from LB-agar plate, pick the colony (mark it on the cap) and stab the colony
- Incubate on shaker overnight
Bacterial Cell Culture Solutions
LB media
- For 1L of LB media add:
- 10g Tryptone
- 10g NaCl
- 5g Yeast extract
- Fill to 1L with DI water
- Autoclave at P6 setting
LB-Agar Plate media
- For 1L of LB-Agar media, mix:
- 960mL water (860 mL if adding glucose)
- 10g Tryptone
- 10g NaCl
- 5g Yeast extract
- 15g Agar (add agar last)
- Autoclave (liquid option): 30 min. sterilization / 20 min drying
- Cool to 50˚C in water bath
- Next three steps are under flame
- (if adding glucose: add 100mL of 20% glucose, final conc. 2% glucose)
- Add antibiotics to the specified final conc:
- Chloramphenicol (CM): 50 µg/mL
- Ampicillin: 100 µg/mL
- Tetracycline: 50 µg/mL
- Kan: 50 µg/mL
- Pour onto the petri dishes
- Mark petri dishes using the following key:
- Ampicillin: one black line
- Chloramphenicol: two black lines
- Kan: one blue line
- Glucose: one red line
Chemically Competent Cells Transformation
- NEB Turbo Chemically Competent Cells (E. coli High Efficiency)
- Thaw chemically competent cells on ice.
- Transfer 50 µl of competent cells to a 1.5 ml microcentrifuge tube (if necessary).
- Add 2 µl of assembled product to NEB competent cells.
- Mix gently by pipetting up and down or flicking the tube 4-5 times. Do not vortex. Place the mixture on ice for 30 minutes. Do not mix.
- Heat shock at 42°C for 30 seconds. Do not mix.
- Transfer tubes on ice for 2 minutes.
- Add 950 µl of room temperature SOC media to tubes.
- Place the tube at 37°C for 60 minutes. Shake vigorously (250 rpm) or rotate.
- Warm selection plates to 37°C.
- Spread 100 µl of the cells onto the plates with appropriate antibiotics. Use amp plates for positive control sample.
- Incubate plates overnight at 37°C.
- SHuffle Express Competent E. coli (C3028)
- Thaw a tube of SHuffle Competent E. coli cells on ice for 10 minutes.
- Add 1–5 µl containing 1 pg–100 ng of plasmid DNA to the cell mixture. Carefully flick the tube 4–5 times to mix cells and DNA. Do not vortex.
- Place the mixture on ice for 30 minutes. Do not mix.
- Heat shock at exactly 42°C for exactly 30 seconds. Do not mix.
- Place on ice for 5 minutes. Do not mix.
- Pipette 950 µl of room temperature SOC into the mixture.
- Place at 30°C for 60 minutes. Shake vigorously (250 rpm) or rotate.
- Warm selection plates to 30°C.
- Mix the cells thoroughly by flicking the tube and inverting, then perform several 10-fold serial dilutions in SOC.
- Spread 50–100 µl of each dilution onto a selection plate and incubate overnight at 30°C. Alternatively, incubate at 25°C for 48 hours.
Qiagen MiniPrep
For overnight cell cultures 1~5mL of E. Coli in LB medium:
- Transfer cell culture into centrifuge tubes
- Centrifuge for 5 min. at 4,000 rpm
- Discard the supernatant
- Resuspend pelleted bacterial cells in 250 µl Buffer P1 and transfer to a microcentrifuge tube. Ensure that RNase A has been added to Buffer P1. No cell clumps should be visible after resuspension of the pellet.
- Add 250 µl Buffer P2 and mix thoroughly by inverting the tube 4–6 times. Do not vortex. Continue inverting the tube until the solution becomes viscous + slightly clear blue. Do not allow lysis reaction to proceed for more than 5 min.
- Add 350 µl Buffer N3 and mix immediately + thoroughly by 4-6x inverting the tube. The solution should become cloudy.
- Centrifuge for 10 min at 13,000 rpm (~17,900 x g) in a table-top microcentrifuge.
- Apply the supernatants from the previous step to the QIAprep spin column by decanting or pipetting.
- Centrifuge for 60 s. Discard the flow-through.
- Wash QIAprep spin column by adding 750µL Buffer PE and centrifuging for 60 s.
- Discard the flow-through, and centrifuge for an additional 1 min to remove residual wash buffer.
- Place the QIAprep column in a clean 1.5 ml microcentrifuge tube. To elute DNA, add 50 µl Buffer EB (10 mM Tris·Cl, pH 8.5) or water to the center of each QIAprep spin column, let stand for 1 min, and centrifuge for 1 min.
Glycerol Stock
- In CryoTube vials, add:
- 250 µL of 50% glycerol
- 250 µL of cell culture
- Store in -80˚C freezer
Protein Protocols
BIO-RAD Protein Gel
- Loading dye + DTT (if not present): in 1.5mL microcentrifuge tube, make (loading dye+DTT) by:
- Get loading dye (Laemmli Sampling Buffer in 203-14A middle drawer under ``Western Blotting Supplies")
- Get DTT (in the biology section of the desiccator in the fridge--which is on the second shelf from bottom right side).
- Mix 54mg of DTT in 950µL of loading dye.
- Determine amount of protein and buffer in each lane according to following table: Note that the protein amount CANNOT exceed 8µg per lane for 10 lane gel, and 6µg per lane for 15 lane gel. Get 1.5mL microtubes as many as needed.
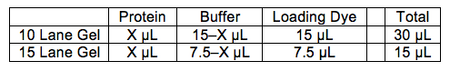
Gel loading conditions - Heat the tubes for 5-10 min, and get the ladder (marked "P" at the top in the Alaska fridge, in the box "Joshi Gel ladders/dyes") out to thaw.
- While the tubes are being heated, prepare the gel:
- Take the green cap off the wells and clean the wells with water. Repeat washing about three times. Take the tape off bottom.
- Assemble the cassette with two gels or (one gel + a gel-wall). Note that the wells should face inwards.
- Put the cassette into the gel box and pour NEW Tris-gly-SDS buffer into the cassette past. The buffer level should be past the wells. Check for any leaking. If there is a leak, reassemble the cassette and try again.
- Set up the voltage machine to the desired voltage and time. For a fast run, 190V with 35 minutes could work. For slow and cleaner result, 90V with 50min could work.
- Now get the heated samples, and the ladder. Add 10µL of ladder for 15 lane gels, 15µL of ladder for 10 lane gels. Add 15µL of the sample for 15 lane gels, 30µL for of the 10 lane gels.
- Fill the box to the 2-gel mark (or 4-gel mark if running more than 3 gels) or more with OLD Tris-gly-SDS buffer. Put the cap on and hit the run button. Check for bubbles rising.

Coomassie Staining
- Retrieve the gel; be careful not to rip, and if sticky apply water. Put it in an empty box.
- Check the staining solution (50% methanol, 40% water, 10% acetic acid, 0.25mg per 100mL) in the fume-hood in the back corridor.
- If the caps have been open, or the solution is out, make a new one (500mL) by:
- 50% of total volume is methanol (is in the flammable cabinet in the chemistry room). 40% of total volume is water. Use plastic graduated cylinder for these.
- 10% of total volume is acetic acid (is in the acid cabinet). BE CAUTIOUS WHEN WALKING CORNERS, and switch gloves after touching acid. Use GLASS graduated cylinder for this.
- measure out and add the Coomassie to appropriate amount. It is located in the "Bahamas"
- NOTE: methanol waste must go to the special waste bin. If the waste bin is full, call the number.
- Pour the Coomassie staining solution into the box for ~20 min.
- Wash the stain with 40% methanol, 10% acetic acid, 50% water for ~30 min.
BLACaM Assays
- Set up plate reader
- Plate should be read at 405 nm
- Shake plate for 2-3 s before reading
- Calculate concentrations and amounts needed to reach a total reaction volume of 150 μL
- Possible conditions to vary
- Ca amount
- EGTA amount (removes Ca)
- Peptide type and concentration
- Other small molecule interactors
- Possible conditions to vary
- Dilute peptides down to working stock concentration (1.25 μM) with 20 mM HEPES buffer (should be room temp)
- Dilute working stock of peptides into multiple tubes to create range of concentrations required by assay
- Note that only 50 μL of the 150 μL well volume will be the peptide, so any concentration will be further diluted by 3x
- Usually no peptide is over 500 nM final concentration in well
- Dilute BlaCaM stock with 20 mM HEPES buffer
- Note that only 20 μL of 150 μL well volume is BlaCaM stock, so final concentration will be 2/15 of diluted stock
- Final concentration of BlaCaM in well should be 125 nM
- Add CaCl2 to 20 mM HEPES to create 10mM Ca concentration
- Plate mixtures in order onto mixing plate (buffer, CaCl2, EGTA, peptides, other molecules, BlaCaM)
- Must use non-binding plates (Corning 3641) for both mixing and assay plates to prevent binding of peptides and BlaCaM to plate
- 75.5 μL 20 mM HEPES + 10 mM Ca to all wells (5 mM final concentration)
- 50 μL 20 mM HEPES (no Ca) to BlaCaM only controls
- 70 μL 20 mM HEPES (no Ca) to blank (no BlaCaM, no peptide) controls
- 50 μL peptide to all but control wells
- Each concentration of peptide should be assayed at least three times
- 20 μL BlaCaM to all wells but blank controls
- Mix each well and incubate for 60 min on shaker (22-24°C)
- At 50 min prepare assay plate by adding 2-3 μL CENTA substrate
- Aim for final CENTA concentration of ~150 μM
- Add 97-98 μL of incubated mixture to assay plate and mix well
- Creates a total assay plate volume of 100 μL
- If possible, use Liquidator pipette system so all 96 wells can be transferred at once
- Immediately take read in plate reader and take reads every 2-3 min for 20 min
GLucCaM Expression
- GLucCaM expression in NEB Turbo E. coli.
- Prepare 5-10mL starter culture from a fresh colony or glycerol stock in LB media supplemented with 50µg/mL CM and 0.2% glucose. Incubate at 37˚C overnight in shaker (225 rpm).
- Next day start 250mL. In 1L baffled flask, add:
- 250mL LB
- 367.5µL of CM (final conc 50µg/mL)
- 2.5mL of the cell culture grown overnight (final 1:100 dilution)
- NO GLUCOSE
- Shake in 37˚C at 225 rpm for about 3 hours to an O.D. of ~0.6
- Induce cells with 0.4mM IPTG and place immediately into an 18˚C incubator for expression. Incubate for 14-18 hours (overnight)
- Spin down cells in large floor centrifuge for 20 mins at 3000g and 4˚C
- Save cells in -20˚C freezer until ready for purification.
- Resuspend cells in lysis buffer (50 mM Tris-CL, pH 7.4 with 300 mM NaCl, 20 mM imidazole)
- Lyse by sonication with 30 second pulse x 3.
- Aliquot the lysed solution into 2mL microcentrifuge tubes and centrifuge at max. rpm for 45 min.
GLucCaM / GLuc Assay
- Prepare the GLuc solution (e.g. 100 samples) by adding 50 µl of BioLux GLuc Flex Substrate and 0.8 ml of BioLux GLuc Flex Stabilizer to 5 ml of BioLux GLuc Flex Assay Buffer (Be sure to prepare enough assay solution as needed for all samples as well as for priming a particular luminometer as recommended by the manufacturer).
- Mix well by inverting the tube several times (Do not vortex).
- Incubate at room temperature for 25 minutes (protect from light in a tightly capped tube/bottle).
- Set the luminometer with the following parameters: 50 µl of injection, 35-40 seconds of delay, and 2-10 seconds of integration.
- Pipet samples (20 µL) into a 96-well white (opaque) or black plate.
- Prime the injector with the assay solution and proceed with the measurement.
Purification of 6xHis-tagged Proteins with Ni-NTA Magnetic Agarose Beads
- Resuspend the Ni-NTA Magnetic Agarose Beads by vortexing for 2 s and then immediately add 200 µl of the 5% Ni-NTA Magnetic Agarose Bead suspension to 1 ml of the lysate containing the 6xHis-tagged protein.
- Mix the suspension gently on an end-over-end shaker for 30 min to 1 h at room temperature (15 - 25°C). It may be necessary to incubate at 4°C if the protein is not stable at room temperature.
- Place the tube on a magnetic separator for 1 min and remove supernatant with a pipet.
- Remove tube from the magnet, add 500 µl of wash buffer, mix the suspension, place the tube on a magnetic separator for 1 min, and remove wash buffer.
- Repeat step 4 another 1-2 times. buffer remaining after the final wash should be removed completely.
- Add 100 µl of elution buffer, mix the suspension, incubate the tube for 1 min, place for 1 min on magnetic separator, and collect the eluate.
- Repeat step 6.
BlaCaM Library Assay
- Use small-volume, nonbinding plates for easiest/best results
- Prep stocks for the final total concentrations/volumes
- Total reaction volume: 60μL
- Peptide concentration: 500nM
- Ca2+ concentration: 5mM
- Centa concentration: 150uM
- Let all reagents adjust to room temperature before adding
- Mix 20mM HEPES buffer with peptide and calcium and put into wells
- Add protein and allow mix to incubate for 1h at RT
- Add Centa substrate, preferably with multichannel/liquidator
- Read absorbance at 405nm after 15min, then every hour until 3 hr
- Watch for color change, if the reaction is proceeding quicker than expected it may be necessary to compress reading intervals
DNA Protocols
Agilent Herculase II PCR Amplification
The following protocol uses Agilent Herculase II as the DNA polymerase for polymerase chain reaction (PCR) amplification of DNA.
- In a PCR tube, mix (50 µL final volume):
- 35 µL water
- 10 µL HercII Buffer (5X)
- 0.5 µL dNTP (25mM each, 100mM total final)
- 1.25 µL forward primers (10 µM)
- 1.25 µL reverse primers (10 µM)
- 1 µL Template DNA (diluted to 10 ng/µL)
- 1 µL HercII DNA polymerase
- Give the tubes a brief spin
- Thermocycling conditions (see diagram below):
- Step 1: 95˚C, 2:00
- Step 2 X30
- Phase 1: 95˚C, 0:15
- Phase 2: (Tm - 5)˚C, 0:20
- Phase 3: 72˚C, 0:30 per 1kb
- Step 3: 72˚C, 3:00

Agilent GeneMorph II Error-Prone PCR
The following protocol uses the Gene Morph II Random Mutagenesis Kit with the Mutazyme II II DNA polymerase for error-prone PCR to randomly mutate a gene for library creation.
- Using the table below, determine the amount of template DNA needed. Keep in mind that the table refers to the amount of target DNA only, not the total amount of plasmid template DNA.
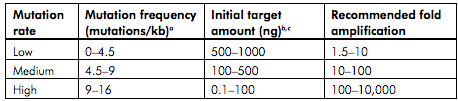
- In a PCR tube mix the following (50 µl final volume)
- 41.5 µl water
- 5 µl of 10x Mutazyme II reaction buffer
- 1 µl of 40 mM dNTP mix (200 µM each final)
- 0.5 µl of primer mix (250 ng/µl of each primer)
- 1 µl of Mutazyme II DNA polymerase (2.5 U/µl)
- 1 µl template (according to below table)
- Centrifuge each reaction briefly.
- Place each reaction in a temperature cycler and run the PCR program described in the diagram below.


Primer Prep
- First dilution: dilute to 100 µM (shortcut: amount_in_nMoles X10 µL)
- Second dilution: dilute to 10 µM
Gibson Assembly
For 2-3 fragment assembly
- Set up the following on ice in PCR tube (total volume 20µL):
- 0.02–0.5 pmols of fragments: 50-100ng of vectors with 2-3 fold of excess inserts.
- 10 µL of Gibson Assembly Master Mix (2X)
- Fill to 20µL with DI water
- Incubate in a thermocycler at 50˚C for 60 min.
NanoDrop
- Wipe the lower and upper measurement pedestals with 2µL DI water
- Measure blank by pipetting 2µL of sample solvent (usually water or DNA elution buffer) on the lower pedestal
- Measure conc. of samples by 2µL on the lower pedestal
ZYMO DNA Clean & Concentrator
ZYMO DNA Clean & Concentrator - 5 Kit was used to purify DNA from PCR.
Protocol:
- Add 2-7 Volumes of DNA Binding Buffer to each volume of DNA sample.
- For plasmid or genomic DNA 2kb, add 2 volumes.
- For PCR or short DNA fragments, add 5 volumes.
- For ssDNA purification, add 7 volumes
- Load the mixture into a Zymo-Spin Column in a Collection Tube.
- Centrifuge at full speed (≥10,000 x g) for 30 seconds. Discard flow through.
- Add 200 µl of DNA Wash Buffer to the column and centrifuge for 30 seconds.
- Repeat previous step, this time centrifuging for 1 minute.
- Place the Zymo-Spin Column into a new 1.5 ml tube. Add ≥6 µl of DNA Elution Buffer or water directly to the column matrix and spin (30 seconds) to elute the DNA.
GeneWiz Sequencing Prep
The following protocol should be followed to prepare DNA for submission to GeneWiz for sequencing.
- Complete online order form.
- Label PCR tubes with the appropriate names form the order form print out.
- Concentrate template DNA according to the table below.
- Mix 10 µl of DNA template with 5 µl of primer.
- Attach PCR tubes to order form and submit GeneWiz mailbox.

