Endy:Double stranding oligo libraries: Difference between revisions
From OpenWetWare
Jump to navigationJump to search
| (11 intermediate revisions by the same user not shown) | |||
| Line 1: | Line 1: | ||
==Order oligos and double-stranding [[Designing_primers|primers]]== | ==Order oligos and double-stranding [[Designing_primers|primers]]== | ||
* Dilute stocks to 100uM | * Dilute stocks to 100uM | ||
* Dilute sequencing primers to 3.2uM (6.4uL of stock solution in 193.6uL water) | * Dilute working stocks of libraries and double-stranding primers to 10uM | ||
* Dilute working stocks of sequencing primers to 3.2uM (6.4uL of stock solution in 193.6uL water) | |||
* Some considerations: | * Some considerations: | ||
| Line 11: | Line 11: | ||
==Double strand the library with modified PCR== | ==Double strand the library with modified PCR== | ||
* | *Expected max library size is 10<sup>8</sup> molecules (limit set by transformation efficiency.) You want to load 10X the expected library size for a single library construction. Therefore, you would like to have 10<sup>9</sup> molecules for a single transformation. | ||
**1pmol corresponds to ~10<sup>11</sup> molecules | |||
* | **Use 25pmol of library to make enough for 2500 transformations | ||
*Total library DNA should be less than ~25pmol per 100uL reaction | |||
===Reaction Mix (100uL)=== | ===Reaction Mix (100uL, 25pmol library)=== | ||
Use the following reaction mix for each PCR reaction: | Use the following reaction mix for each PCR reaction: | ||
*10 μl 10x Thermo polymerase buffer | *10 μl 10x Thermo polymerase buffer | ||
| Line 26: | Line 27: | ||
===PCR protocol=== | ===PCR protocol=== | ||
*95 C for 2.5 minutes | *95<sup>o</sup>C for 2.5 minutes | ||
*Cycle 5 times: | *Cycle 5 times: | ||
**55 C (or whatever temperature is appropriate) for 30 | **55<sup>o</sup>C (or whatever temperature is appropriate) for 30 seconds (annealing) | ||
**72 C for 1.5 minutes (elongation) | **72<sup>o</sup>C for 1.5 minutes (elongation) | ||
*72 C for 10 minutes (final elongation) | *72<sup>o</sup>C for 10 minutes (final elongation) | ||
*4 C forever | *4<sup>o</sup>C forever | ||
==PCR cleanup on the double-stranded | ==Perform PCR cleanup on the double-stranded library== | ||
* This concentrates the samples and allows for the buffer to be switched to something more appropriate. | * This concentrates the samples and allows for the buffer to be switched to something more appropriate. | ||
* PCR purification columns can handle up to 10ug of DNA | * PCR purification columns can handle up to 10ug of DNA | ||
** 100pmol of a 100bp oligo is about 3ug, so multiple 100-ul reactions of 25pmol can be combined into one column | |||
* Expected recovery from a PCR purification reaction is 90% (from the Invitrogen package) | * Expected recovery from a PCR purification reaction is 90% (from the Invitrogen package) | ||
* You can run a sample of the PCR product out on a gel against a sample of the original library to verify that the double stranding worked (double stranded DNA should run slightly faster than single stranded) [[Image:Double-stranded_oligo_libraries.jpg|thumb|none|300px|Three libraries ~100bp; on the left is the single-stranded oligo; on the right are double-stranded oligos (different lanes are different primers)]] | |||
==[[Restriction digest]] the | ==[[Restriction digest]] the library== | ||
== | ==Perform PCR cleanup on the digest== | ||
* | * This will remove the cut ends, since they are small. | ||
==[[DNA_Ligation|Ligate]] the sample from the PCR cleanup with a vector== | ==[[DNA_Ligation|Ligate]] the sample from the PCR cleanup with a vector== | ||
| Line 47: | Line 50: | ||
==Transform into compotent cells== | ==Transform into compotent cells== | ||
* This will either be done via [[Electroporation|electroporation]] or [[Transforming_chemically_competent_cells|chemically compotent cells]], we’re experimenting now to see which one is more efficient. | * This will either be done via [[Electroporation|electroporation]] or [[Transforming_chemically_competent_cells|chemically compotent cells]], we’re experimenting now to see which one is more efficient. | ||
[[Category:Protocol]] | [[Category:Protocol]] | ||
[[Category:DNA]] | [[Category:DNA]] | ||
[[Category:In vitro]] | [[Category:In vitro]] | ||
Latest revision as of 19:39, 29 August 2006
Order oligos and double-stranding primers
- Dilute stocks to 100uM
- Dilute working stocks of libraries and double-stranding primers to 10uM
- Dilute working stocks of sequencing primers to 3.2uM (6.4uL of stock solution in 193.6uL water)
- Some considerations:
- Oligos should be the maximum length because this will help with PCR cleanup and ligation efficiency
- Make sure you have some spacer sequence around the restriction site. NEB has a list of the length of the spacer sequence required for each restriction enzyme. (8bp is usually a safe bet)
- Order the lowest concentration allowable for the size oligo you want – this will be 50nmole for the 100bp oligo. This will already be more than you’ll need.
- If you don’t mind spending more money you can order special “doped” oligo pools where instead of even concentrations of A/T or A/T/C/G or A/T/C, you get 90%A/2%C/8%G, etc. This allows for you to generate a library which is much more likely to produce productive clones.
Double strand the library with modified PCR
- Expected max library size is 108 molecules (limit set by transformation efficiency.) You want to load 10X the expected library size for a single library construction. Therefore, you would like to have 109 molecules for a single transformation.
- 1pmol corresponds to ~1011 molecules
- Use 25pmol of library to make enough for 2500 transformations
- Total library DNA should be less than ~25pmol per 100uL reaction
Reaction Mix (100uL, 25pmol library)
Use the following reaction mix for each PCR reaction:
- 10 μl 10x Thermo polymerase buffer
- 10 μl 10x dNTPs (10x = 2.5 mM each dNTP)
- 5 μl 10 μM FWD primer
- 5 μl 10 μM REV primer
- 1 μl Polymerase (taq or vent)
- 66.5 μl H2O
- 2.5 μl 10μM library stock
PCR protocol
- 95oC for 2.5 minutes
- Cycle 5 times:
- 55oC (or whatever temperature is appropriate) for 30 seconds (annealing)
- 72oC for 1.5 minutes (elongation)
- 72oC for 10 minutes (final elongation)
- 4oC forever
Perform PCR cleanup on the double-stranded library
- This concentrates the samples and allows for the buffer to be switched to something more appropriate.
- PCR purification columns can handle up to 10ug of DNA
- 100pmol of a 100bp oligo is about 3ug, so multiple 100-ul reactions of 25pmol can be combined into one column
- Expected recovery from a PCR purification reaction is 90% (from the Invitrogen package)
- You can run a sample of the PCR product out on a gel against a sample of the original library to verify that the double stranding worked (double stranded DNA should run slightly faster than single stranded)
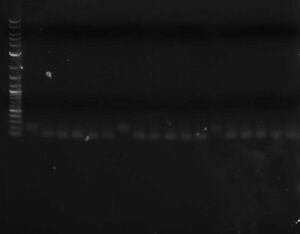
Three libraries ~100bp; on the left is the single-stranded oligo; on the right are double-stranded oligos (different lanes are different primers)

Restriction digest the library
Perform PCR cleanup on the digest
- This will remove the cut ends, since they are small.
Ligate the sample from the PCR cleanup with a vector
Transform into compotent cells
- This will either be done via electroporation or chemically compotent cells, we’re experimenting now to see which one is more efficient.
