20.109(S11):Initiating transcript and protein assays (Day5)
_frontpg.JPG)
Introduction
There are several ways to assess protein concentration, either quantitatively or as present above some threshold value. In the second module, you used an enzymatic assay to detect a specific protein, β-galactosidase. However, not all proteins have straightforward functional assays that can be developed for their detection. A great universal way to identify a specific protein from a complex mixture is to exploit antibodies – also called immunoglobulins – whether in a Western blot or by ELISA (enzyme-linked immunosorbent assay). In native physiological settings (such as your own body), antibodies are secreted by B cells in response to pathogens. A given antibody is highly specific ([math]\displaystyle{ K_D }[/math] ~ nM) for its binding partner, called an antigen, and the entire antibody population for a given person is incredibly diverse (>107 unique antibodies). Diversity is maintained by recombination processes at the DNA level, and specificity entailed by protein structure.
Antibody proteins comprise constant (C) and variable (V) regions, on both their heavy and light chains. The C regions determine antibody effector functions, such as antibody-dependent killing of infected cells. The three hypervariable portions of the V region together make up the antigen-recognition site. Only a small portion of an antigen, called an epitope, is recognized by its cognate antibody. This ~10 amino acid region may be linear, or it may be made up of linearly distant regions and thus recognized only when the antigen is in its native conformation. For example, conformation-dependent antibodies are useful for distinguishing different collagen types.

Antibodies can be raised in animals or special cell lines and even be genetically engineered. Polyclonal antibodies (pools of antibodies that recognize distinct epitopes on the same antigen) can be obtained from animal serum. The animal is infected with the antigen of interest in the presence of a costimulatory signal, usually multiple times, and then bled. In this case, a large fraction of the antibodies obtained will not be against the antigen of interest. In contrast, monoclonal antibodies can be made both highly specific and pure. In this process, normal antibody-producing B cells are fused with immortalized B cells derived from myelomas, and the two cell types are fused by chemical treatment with a limited efficiency. To select only heterogeneously fused cells, the cultures are maintained in medium in which myeloma cells alone cannot survive (often HAT medium). Normal B cells will naturally die out over time with no intervention, so ultimately only the fused cells, called hybridomas, remain. Genetic engineering can be used to combine a human antibody ‘frame’ (all of the C and part of the V region) with an antigen-recognition site discovered in another species (e.g., murine). When antibodies are used as therapeutics, this decreases the possibility that the patient’s body will treat them as foreign, compared to an antibody produced from only mouse genes. Normally, injecting an antibody from species X into an animal of species Y will cause production of anti-X antibodies, called secondary antibodies. These can be very useful in technical assays, as you will see below.
Today you will use antibodies against collagen in an indirect ELISA assay. Both indirect and sandwich ELISA are shown in the figure at right – can you see why sandwich ELISA might be the superior assay with respect to sensitivity and specificity? In indirect ELISA, the first step is to bind protein extracts, obtained from your two different culture conditions, to well plates. Next you will add a primary antibody that recognizes a particular antigen – namely, epitopes on collagen I or collagen II – to the relevant wells. (Actually, before adding the antibody you will "block" the plate with milk protein to prevent non-specific binding of the antibody.) Next, any excess antibody must be washed away with a mild detergent. Finally, a secondary antibody – namely one that recognizes the primary antibody – must be added. The secondary antibody is conjugated to alkaline phosphatase, which will undergo a colorimetric reaction in the presence of its substrate. Thus, the relative quantity of protein can be assessed by absorbance spectroscopy following substrate addition. To quantify the absolute amount of protein, you will run dilutions of a collagen standard in parallel with your culture samples.
During your ELISA incubation steps, you will prepare three qPCR reactions for each cDNA mixture that you prepared last time. The first two reactions will use primers for our genes of interest, namely collagen II and collagen I. The final reaction will be a control for cDNA amount that contains primers for 18S ribosomal RNA. The culture condition that has relatively higher collagen II and relatively lower collagen I, taking into account amount loaded based on 18S results, should be the culture with more chondrocyte-like cells.
Reference: Abbas, A.K. & Lichtman, A.H. (2005). Cellular and Molecular Immunology (5th ed.). Philedelphia: Elsevier Saunders.
Protocols
Part 1: Day 1 of ELISA
ELISA protocol
We will run this assay in a 96-well microtiter plate and make measurements using an absorbance plate reader, just as we did for the Miller assays in Module 2.
- Spin your pepsin-digested samples from last time in the microfuge for 5 minutes at maximum speed.
- Label one 96-well plate for your collagen I assay, and one for your collagen II assay.
- Separate plates will make it convenient to keep track of antibody addition and to later have different reaction stop times if necessary.
- The first step in indirect ELISA is to adsorb all your samples to the wells. You will also need to prepare standard samples in the same plate, which get treated just the same as your test samples. These standards will be used as a reference for protein concentration. Both standards and unknown samples will be run in duplicate (see figure at right).
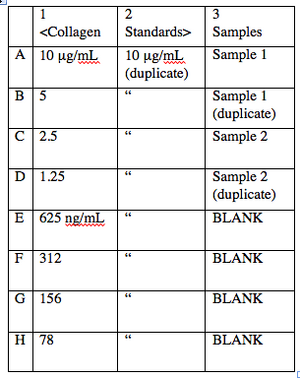
Suggested ELISA plan. This plan can be used for both your collagen I and your collagen II plate. In each case, columns 1 and 2 are duplicates of the collagen standards, and column 3 contains your experimental samples and a few wells (labeled BLANK) to measure background. - You will be given 250 μL aliquots of collagens I and II at 10 μg/mL each. Prepare your standards as follows:
- Ultimately, you want to pipet 50 μL per well of each standard concentration, two wells per standard.
- Option 1:
- Prepare 7 eppendorf tubes with 120 μL of PBS each.
- Add 120 μL of the 10 μg/mL collagen to the first eppendorf tube, and vortex.
- Now take 120 μL of that standard (now 5 μg/mL) and add it to the next eppendorf tube.
- When you are all done or as you go, pipet the standards into the appropriate wells.
- Option 2:
- Pipet 50 μL of PBS into wells 1 and 2 of rows B-H (skip A!).
- Pipet 100 μL of the 10 μg/mL collagen into the appropriate wells (A1 and A2).
- Using a regular or multichannel pipet, transfer 50 μL of these solutions to the next wells down (B1 and B2), and mix with the PBS.
- Repeat, now moving 50 μL of the 5 μg/mL solution in the B wells down to the C wells.
- Either of these methods is called making doubling dilutions. Which way do you think introduces less error?
- Now add 50 μL of your samples to the appropriate wells. For the blank wells you should add PBS.
- Cover each plate (CN I and CNII) when you are done, wrap around it with parafilm to better prevent evaporation, and allow the samples to sit for 80 minutes. In the meantime, set up your qPCR reactions (Part 2).
- After the incubation time has passed, you will wash and then block your plate.
- First, flick the solutions in the plate into the sink.
- Using the multichannel pipet and a reservoir, add 200 μL of Wash Buffer to each well, then gently swirl the plate (by hand) for a few seconds.
- Flick the solutions out again, and then blot the plate against paper towels. You can smack the plates pretty hard, but it is possible to break them!
- Repeat the wash one more time.
- Finally, add 200 μL of Block Buffer to the plate. Wait another 60-90 minutes. In the meantime, you can work on cell viability analysis (first listed on Day 4 Part 4).
- Repeat the wash step that you performed above, again with two rinses.
- When you are ready, ask the teaching faculty for some primary anti-collagen antibodies (these should be diluted at the last minute). Add 100 μL of diluted antibody per well.
- Your samples will be left overnight in antibody solution, then moved back to block buffer by the teaching faculty.

Part 2: Set up qPCR reaction
Important background, do not skip
Each group will have 6 samples in duplicate, or 12 reactions in total. In the schematic below, the top row indicates the group colour (obviously!). The next row indicates the sample identity as X-Y, where X is the culture condition (keep track of what is 1 and 2) and Y is the replicate number. The final row indicates the gene of interest and thus the primer set that will be used.
Ultimately, you will add your cDNAs to the appropriate wells in your row. Then, the teaching faculty will add the appropriate master mix (containing water, primers, SYBR green dye, buffer, etc.) to all of the wells with a multi-channel pipet. We are doing most of the preparations for you today for a few reasons. For the master mixes, qPCR is very sensitive, so we want to reduce error and any differences between samples as much as possible. For the plating, note that the DNA-binding dye is very light-sensitive, so we want to do this step quickly rather than one group at a time. (Similar reasoning about reducing pipetting variability in the plating step also applies.)

Preparation
- For each culture condition, you need to prepare enough diluted cDNA for 6 reactions, plus some extra. Unless you had really low RNA yield (talk to the teaching faculty if so), you should prepare the following as your "1x" stock of cDNA, times 7 reactions: 1.9 μL of cDNA plus 3.1 μL of water (so 13.3 plus 21.7 μL, respectively).
- We normally prepare qPCR reactions with slightly different volumes than we'll ask you to use today (long story...), so for my ease of comparison later as I revise the module and see what works well, you will consider the above cDNA 1x or undiluted stock.
- The primer sets and cycling conditions have been validated for a wide range of starting cDNA concentration. Decide what you want to use according to the criteria below, and prepare 30 μL plus at least 10-20% excess of your dilution (in nuclease free water).
- If you had 500 ng (or close to it) of RNA in your cDNA reaction, a 5-10x further dilution of your cDNA should work well.
- If you had a bit less, using the stock directly or a 2x dilution may be more appropriate.
- Add 5 μL of your cDNAs to your 12 wells according to the schematic above and check your group colour off the list.
- When everyone is done, the teaching faculty will add the three different master mixes, and run the plate over to the [BioMicroCenter | BioMicro center]] shared equipment facility in building 68.
- Groups that happen to have down-time when the plate is ready may come and observe the plate set-up.
Part 3: Continue viability analysis (optional)
Assuming you didn't have a chance to finish (or even start!) the viability analysis last time, see Day 4 Part 4 and hand this work in either today or next time.
For next time
There will be no more assignments to hand in besides the two remaining major assessments. However, by the end of Day 6 or so (whether in or outside class), you should meet with someone from another group to discuss your research proposal. See Part 4 of Day 6 for a description of how to proceed and the Day 6 Talk page for the randomized list of pairings. (You and your partner should meet separately with people who are not themselves in the same group, so that you each get a chance to give your take on the project.)
Reagent list
- qPCR reagents
- 2X Power SYBR Green Master Mix from Applied Biosystems
- Collagen II primers, 100 nM
- Collagen I primers, 100 nM
- 18S primers, 25 nM
- ELISA solutions
- PBS reconstituted from EMD tablets
- 140 mM NaCl
- 10 mM phosphate buffer
- 3 mM KCl
- pH 7.4
- Wash buffer
- PBS
- 0.05 % Tween 20
- Block buffer
- PBS
- 5 % powdered milk
- PBS reconstituted from EMD tablets
- ELISA collagen-specific reagents
- All from GeneTex
- Collagen I standard, diluted from 1 mg/mL in PBS
- Collagen II standard, diluted from 1 mg/mL in PBS
- Collagen I antibody, diluted 1:4000 from 1 mg/mL in PBS
- Collagen II antibody, diluted 1:4000 from 1 mg/mL in PBS
