20.109(S08):Induce protein expression (Day4)

Introduction

.png)
Last time you transformed your mutant DNA into BL21(DE3) cells. The colonies that arose were moved to liquid cultures, and today you will add IPTG to these cultures to induce protein expression by the bacteria. Next time you will purify the resultant protein. I won’t shy away from telling you that there are many things that can go wrong at this stage! However, each one is certainly a learning experience.
As evidenced by Nagai’s work, wild-type inverse pericam is not toxic to BL21(DE3) cells. Although it is unlikely for your small mutation to dramatically change this fact, in general a novel protein may turn out to be toxic. If this is the case, only very small amounts of protein are produced before the bacteria die. Keep in mind that overexpressing a single protein may come at the expense of producing proteins needed for survival, and will most likely cause cell death eventually; however, toxic proteins hasten this demise. Aberrant toxicity can sometimes be alleviated by reducing the culture temperature (e.g., to 30 °C).
Based on its fluorescence activity, wild-type inverse pericam allows proper folding of (cp)EYFP, and based on its response to calcium, it also allows calmodulin to fold. One problem you may encounter is that your mutant proteins will no longer fold correctly. Since you made mutations in the calcium sensor part of IPC, rather than the fluorescent part, it is unlikely that your protein will destroy EYFP fluorescence. However, a common problem with misfolded proteins is the formation of insoluble aggregates, due for instance to improperly exposed hydrophobic surfaces. Proteins can be purified from these aggregates – called inclusion bodies – but the process is more labour-intensive than for soluble proteins. (The proteins must be extracted under more harsh conditions than you will use next time, then purified under denaturing conditions, before finally attempting to renature the proteins.) Inclusion bodies sometimes form simply due to very high expression of the protein of interest, causing it to pass its solubility limit. This outcome can be prevented by lowering the culture temperature or time, the amount of IPTG, or the growth phase of the bacteria.
One final point to keep in mind is that not all proteins can be produced in bacteria. Eukaryotic proteins that require post-translational modifications (such as glycosylation) for activity require eukaryotic hosts (such as yeast, or the ubiquitous CHO – Chinese hamster ovary – cells). Sometimes eukaryote-derived proteins will be truncated or otherwise mistranslated by E. coli due to differential codon bias; errors in translation can be prevented by providing additional tRNAs to the culture or directly to the bacteria via plasmids. Despite all this complexity, prokaryotic hosts have been plenty good enough to produce proteins for certain therapies, notably the cytokine G-CSF for patients needing to replenish their white blood cells (e.g., after chemotherapy), sold as Neupogen by Amgen.

After you induce your cells with IPTG, you will let the resultant protein factories do their work for 2-3 hours. During this time, you will look at the sequencing results for your mutant plasmids, to determine if they have the right mutation (and only that mutation). The invention of automated sequencing machines has made sequence determination a fast and inexpensive endeavor. The method for sequencing DNA is not new but automation of the process is recent, developed in conjunction with the massive genome sequencing efforts of the 1990s. At the heart of sequencing reactions is chemistry worked out by Fred Sanger in the 1970’s which uses dideoxynucleotides (see figure at right).
These chain-terminating bases can be added to a growing chain of DNA but cannot be further extended. Performing four reactions, each with a different chain-terminating base, generates fragments of different lengths ending at G, A, T, or C. The fragments, once separated by size, reflect the DNA’s sequence. In the “old days” (all of 10 years ago!) radioactive material was incorporated into the elongating DNA fragments so they could be visualized on X-ray film (image on left). More recently fluorescent dyes, one color linked to each dideoxy-base, have been used instead. The four colored fragments can be passed through capillaries to a computer that can read the output and trace the color intensities detected (image on right). Your sample was sequenced in this way on an ABI 3730 DNA Analyzer.
-
 Sequencing gel
Sequencing gel -
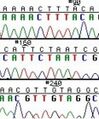 Sequence trace data
Sequence trace data


Analysis of sequence data is no small task. “Sequence gazing” can swallow hours of time with little or no results. There are also many web-based programs to decipher patterns. The nucleotide or its translated protein can be examined in this way. Thanks to the genome sequence information that is now available, a new verb, “to BLAST,” has been coined to describe the comparison of your own sequence to sequences from other organisms. BLAST is an acronym for Basic Local Alignment Search Tool, and can be accessed through the National Center for Biotechnology Information (NCBI) home page.
Protocols
Part 1: Cell measurement and IPTG induction
- For each mutant protein, you will be given a 6 mL aliquot of BL21(DE3) cells carrying the mutant plasmid; you will also receive a tube of BL21(DE3) carrying wild-type inverse pericam. These cells should be in or close to the mid-log phase of growth for good induction, just as they were for transformation. Like last time, check the OD600 value of your cells until it falls between 04.and 06. (Better to overshoot a little than undershoot.)
- Once your cells have reached the appropriate growth phase, set aside - on ice - 1.5 mL of cells from each tube as a control (no IPTG) sample. Then take an aliquot of cold IPTG (0.1 M), and add to your remaining cells at a final concentration of 1 mM. You should prepare four mutant and one wild-type tube.
- Return your tubes to the rotary shaker in the 37 °C incubator, and note down the time.
- While your IPTG-stimulated cells are producing protein, you will analyze the sequence data for the plasmids they are carrying. At the end of the day, you will choose only one colony from each mutant to save, and give the other sample to the teaching faculty to be bleached and thrown away.
Part 2: Analyze sequence data
Your goal today is to analyze the sequencing data for four samples - two independent colonies from each of two mutants - and then decide which colony to proceed with for each mutant. You will want to have this document handy, and mark the expected location of your mutation with bold text before proceeding. (Just compare to your annotation of the old IPC sequence document, using Word Count or the Find tool.) The new file contains the inverse pericam sequence as before, but also the flanking sequences (from the pRSET vector) before and after IPC, which are separated from IPC by a blank line. The second page of the document contains the reverse complement of IPC/pRSET, which was gotten using this website. Be sure to bold your mutant codon in the reverse complement sequence as well.
The data from the MIT Biopolymers Facility is available at this link. Choose the "Login to dnaLIMS" link and then use "astachow" and "be109" to login. At the bottom of the left panel should be a link to download your sequencing results. Select the appropriate order # (you'll be told which one is correct) and then "submit." The quickest way to start working with your data is to follow the "view" link. From this link you'll see the sequencing traces and can click on on "sequence text" to view it.
Rather than look through the sequence to magically find the relevant portion, you can align the data you just got with the standard inverse pericam sequence and the differences will be quickly identified. There are several web-based programs for aligning sequences and still more programs that can be purchased. The steps for using one web-based tool are sketched below.
Align with "bl2seq" from NCBI
- The alignment program can be accessed through the NCBI BLAST page or directly from this link
- To allow for gaps in the sequence alignment, uncheck the "filter" box. All the other default settings should be fine.
- Paste the sequence text from your sequencing run into the "Sequence 1" box. This will now be the "query." If there were ambiguous areas of your sequencing results, these will be listed as "N" rather than "A" "T" "G" or "C" and it's fine to include Ns in the query.
- Paste the inverse pericam sequence into the "Sequence 2" box. For samples probed with the forward primer, use the regular IPC sequence; for those using a reverse primer, you should put in the reverse complement. Which alignment will be more useful depends on the location of your mutation.
- Click on Align. Matches will be shown by vertical lines between the aligned sequences. You should see a long stream of matches, followed by lots of errors in the last ~200bp of the sequence – ignore the error-ridden part of the data, as it may not accurately reflect your mutant plasmid. In this stream of matches, the the 1-3 missing lines indicating your mutant codon should stand out. If they don’t, use the numbering or Find tool to locate the appropriate codon.
- You should print a screenshot of each alignment to pdf (and to paper if you desire). These will be used to prepare a figure showing what you found today. You might want to email yourself the alignment screen shots or post them to your wiki userpage.
If both colonies for a given mutant have the correct sequence, flip a coin and proceed with one or the other; ditto if both are inconclusive. If one appears right and the other doesn’t, of course proceed with the former. Finally, if both are clearly wrong, talk to a member of the teaching faculty.
For your reference, another popular sequence alignment program is "CLUSTAL-W" from EMBL-EBI , located here.
Part 3: Observe mutant colonies
Last time you transformed BL21(DE3) cells with four different plasmids (two mutants, two candidates each). Compare the relative colony formation of cells carrying the different plasmids, at whichever cell concentration (1X or 10X dilution) it is feasible to do so. If all the plates have dense cell growth, there is no need for you to get an exact colony count; just do your best to get a relative estimate, and describe any findings in your notebook.
Part 4: Cell observation and collection
- After ~2 hours, you will pour 1.5 mL from each tube into a labeled eppendorf, then spin for 1 min. at maximum speed. Save the other 3 mL!
- Aspirate the supernatant from each eppendorf, using a fresh yellow pipet tip on the end of the glass pipet each time.
- Observe the colour of each of your pellets, and compare to the examples on today's talk page. If the wild-type and both mutant pellets all appear yellow-greenish to the eye, proceed as follows:
- Do NOT toss the rest of the liquid cultures. First, measure their OD600 values, according to part 5 of today's protocol.
- Next, pour 1.5 mL more of the relevant liquid culture on top of each pellet, spin again, and aspirate the supernatant.
- The last 1.5 mL of culture may be bleached and discarded.
- If one or more of your pellets are white or only dimly coloured, please ask one of the teaching staff too show you the room temperature rotary shaker in the back room. You will continue to grow your bacteria overnight. Tomorrow morning, the teaching staff will collect your pellets for you and freeze them. As you can see above, the +IPTG pellets are from 3 mL of culture, while the -IPTG pellets come from 1.5 mL of culture.
Part 5: Preparation for next time
Next time, you will lyse your bacterial samples to release their proteins, and run these out on a protein gel. In order to compare the amount of protein in the -IPTG versus +IPTG samples, you would like to normalize by the number of cells. At this point, you may have only three samples ready (-IPTG only), or you may have all six. In either case, measure the OD600 of a 1:10 dilution of cells for each finished sample, and write this number down in your notebook. Then spin down the cells and aspirate the supernatant. (Be sure to combine pellets for the +IPTG samples.)
For next time
- Taking into account the comments you received on your outline, write a draft of Part 1 of your portfolio.
- While the results are fresh in your mind, you might also prepare the table(s) and/or sentence(s) that pertain to your colony counts and cell pellet observations, and a figure depicting your sequencing results. This part will not be graded. However, you may turn it in for feedback if you want.
- If the sequencing data was not available in time today, analyze it as part of your homework instead, and come prepared next time with a choice of which samples to purify.
- Please download the midsemester evaluation form found here. Complete the questionnaire and then print it out without including your name to turn in next time. If there is something you'd like to see done differently for the rest of the course, this is your chance to lobby for that change. Similarly, if there is something you think the class has to keep doing, let us know that too.
- Day 5 of this module will be an intense one, and has the potential to run long. You will do yourself a great service if you carefully read the text in advance, then consider what preparations you will need to do on that day, and organize your thoughts in your notebook and/or with your partner. This part will not be collected or evaluated.
- Finally, you might start thinking about your journal club presentation (see Day 8 for list of papers) and portfolio Parts 3 and 4 as well. These will come up faster than you think!
Reagent List
- IPTG (isopropyl β-D-1-thiogalactoside), 0.1M
