20.109(S08):DNA engineering/Restriction map and tissue culture (Day 6)

Introduction
In the past century, we have learned a tremendous amount by studying the behavior of mammalian cells maintained in the laboratory. Tissue culture was originally developed about 100 years ago as a method for learning about mammalian biology. The term tissue culture was coined because people were doing exactly that, extracting tissue and letting it live in a dish for a short time. Today, most tissue culture experiments are done using isolated cells rather than whole tissues. Much of what we know about cancer, heritable diseases, and the effects of the environment on human health has been derived from studies of cultured cells.

What types of cells do people study, and where do they come from? Cells that are isolated from tissue are called primary cells, because they come directly from an animal. It is very difficult to culture primary cells, largely because primary cells that are placed in culture divide only a limited number of times. This limitation on the lifespan of cultured primary cells, called the Hayflick limit, is a problem because it requires a researcher to constantly remove tissues from animals in order to complete a study. Cell isolation processes can be quite labour-intensive, and also can complicate data analysis due to inherent animal-to-animal variation. To get around the first of these problems, people have studied cells that are immortal, which means that they can divide indefinitely. (Some inherent cell-to-cell variation still exists in such cells.)
One familiar type of immortalized cell is the cancer cell. Tumor cells continuously divide, allowing cancer to invade tissues and proliferate. Cancer cells behave the same way in culture, and under the right conditions, cells can be taken from a tumor and divide indefinitely in culture. Another type of immortalized cell is the embryonic stem cell. Embryonic stem cells are derived from an early stage embryo, and these cells are completely undifferentiated and pluripotent, which means that under the right conditions, they can become any mammalian cell type. Mouse embryonic stem cells have become a valuable research tool, and it is this cell type that we will be using for our current experimental module.
The art of tissue culture lies in the ability to create conditions that are similar to what a cell would experience in an animal, namely 37°C and neutral pH. Blood nourishes the cells in an animal, and blood components are used to feed cells in culture. Serum, the cell-free (and clotting-factor free) component of blood, contains many of the factors necessary to support the growth of cells outside the animal. Consequently, serum is frequently added to tissue culture medium, although serum-free media exist and support some types of cultured cells.
Cultured mammalian cells must grow in a germ-free environment and researchers using tissue culture must be skilled in sterile technique. Germs double very quickly relative to mammalian cells. An average mammalian cell doubles about once per day whereas a bacterium is able to double every 20 minutes under optimal conditions. Consequently, if you put 100 mammalian cells and 1 bacteria together in a dish, within 24 hours you would have ~200 unhappy mammalian cells, and about 100 million happy bacteria! Needless to say, you would not find it very useful to continue to study the behavior of your mammalian cells under these conditions!
One major objective for this experimental module is for you to learn how to perform tissue culture. You will learn how to get mouse embryonic stem cells to grow in a dish and also how to prevent contaminants from getting into your cell cultures. All of this will form the foundation for the ultimate experiment, which is to detect DNA rearrangements in mammalian cells by the appearance of a fluorescent signal.
Protocol
The tissue culture facility in the lab can accommodate 6 students at a time. Half the class will begin by performing the tissue culture portion of today’s experiment while the other half will begin in the main lab loading their samples from last lab on an agarose gel.
Part 1: Agarose Gel Electrophoresis
Load your samples on a 1% agarose gel in the following order:
Lane Sample Volume to load 1 Molecular Weight Marker-see Day3 5 ul 2 pCX-NNX diagnostic digest 1 25 ul 3 Candidate 1 diagnostic digest 1 25 ul 4 Candidate 2 diagnostic digest 1 25 ul 5 Candidate 3 diagnostic digest 1 25 ul 6 empty xxx 7 pCX-NNX diagnostic digest 2 25 ul 8 Candidate 1 diagnostic digest 2 25 ul 9 Candidate 2 diagnostic digest 2 25 ul 10 Candidate 3 diagnostic digest 2 25 ul
Once all the samples are loaded, the power will be applied (100V for 45 minutes) and the gel will be photographed. While you are waiting, you can review and discuss the assigned article.
Part 2: Tissue Culture
Each of you will have a 60 mm dish of mouse embryonic stem (MES) cells that you will use to seed a six-well dish. You and your partner will seed the dishes at different concentrations so you should decide who will seed at 1:100 and who will seed at 1:400.
- Prewarm all the required reagents in the water bath.
- Look at your cells as you remove them from the incubator. Look first at the color and clarity of the media. Fresh media is reddish-orange in color and if the media on your cells is yellow or cloudy, it could mean that the cells are overgrown, contaminated or starved for CO2. Next look at the cells on the inverted microscope. Note their shape and arrangement in the dish and how densely the cells cover the surface.
- Move the cells into the sterile hood, as well as the PBS, trypsin, and media that you will need. One of the greatest sources for TC contamination is moving materials in and out of the hood since this disturbs the air flow that maintains the sterile environment inside the hood. Anticipate what you will need during your experiment to avoid moving your arms in and out of the hood while your cells are inside.
- Aspirate the media from the cells.
- Wash the cells by adding 3 ml PBS with a 5 ml pipet. Tip the dish in all directions to rinse all the cells, and then aspirate the liquid out of the dish.
- Repeat the PBS wash, leaving the cells dry for the next step.
- To dislodge the cells from the dish, you will add trypsin, a proteolytic enzyme. Using a 2 ml pipet, add 0.5 ml of trypsin to the dish. For one minute precisely (use your timer), tip the dish in each direction to distribute the trypsin over the cells then aspirate the trypsin off the cells. Incubate the cells (“dry”) at 37° for 10 minutes, again using your timer to precisely time this incubation.
- While you are waiting, you can add 1 ml of gelatin to each well of a six-well dish. This should be done in the sterile hood with sterile technique. The gelatin will be removed before you seed the dish with your MES cells but it is important to pre-treat the dish this way. The gelatin must remain in the wells for at least 10 minutes.
- With a 5 ml pipet, add 4 ml of media to the trypsinized MES cells and pipet the liquid up and down (“triterate”) to remove the cells from the plastic and suspend them in the liquid. Remove a small amount of the liquid to an eppendorf tube and take it to the inverted microscopes.
- Fill one chamber of a hemocytometer with 10 ul of the cell suspension. This slide has an etched grid of nine large squares. The square in the center is further etched into 25 squares each with a volume of 0.1 ul and 16 tiny chambers (4x4 pattern). The concentration of cells in a sample can be determined by counting the cells that fall within the 4x4 pattern and then multiplying by 10,000 to determine the number of cells/ml. You should count the cells in the four corner squares of the 25 square grid, then average the numbers to determine the concentration of cells in your suspension.
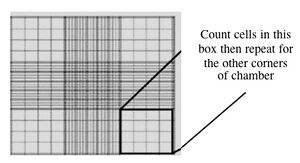
Counting cells using a hemocytometer - You and your partner will seed at different concentrations. Decide if you will try the 1:100 or 1:400 dilution and add the appropriate amount of cell suspension to 10 ml of media in a 15 ml conical tube.
- Remove the gelatin from the six-well dish you have prepared and add 3 ml of your cell dilution to each well. Be sure to label your dish with your name, today’s date, the cell line (called “J1”) and the type of media you have used. Return your cells to the incubator.
- Aspirate any remaining cell suspensions to destroy them and clean up the hood. Dispose any vessels that held cells in the Biohazard waste and any sharps in the grey bins. The next group who uses your hood should find the surfaces wiped down, no equipment left inside, the sash closed and the germicidal lamp on.

DONE!
For next time
- Prepare what will become Figures 5 and 6 in your lab report, including captions. Briefly, Figure 6 shows your diagnostic gel, while Figure 5 is a depiction of your restriction analysis strategy. You may also want to write your Methods sub-section pertaining to diagnostic digests, but this will not be collected.
- Now that you have discussed some primary literature in class, write a draft of the Introduction to your lab report (#4 under Order of Assembly on the lab report page). You need only write the first half at this time, where you set the stage for your experiments by discussing homologous recombination in a general way. For your final draft, you will need to link this discussion to the purpose and significance of your own experiments.
- Fill out the tables found in the protocols section of Module 1 Day 7. It is important that you arrive in lab with these calculations completed.
- Calculate the number of cells in each well of your six well dish. Determine how many cells there should be next time if:
- only 25% of the cells are able to stick and proliferate (this is called a 25% plating efficiency),
- the doubling time for the cells is 24 hours, and
- the cells take 24 hours to recover from trypsin treatment before they begin doubling.
Reagents list
- Trypsin
- 0.25%Trypsin
- 1 mM EDTA
- PBS
- Dulbecco’s Phosphate-Buffered Saline
- J1 culture medium
- 100 U/ml Pen/Strep
- 0.3 mg/ml glutamine
- 0.1 mM BME,
- 1 mM Nonessential amino acids
- 10% serum
- LIF
- Gelatin
- 0.1% TC-grade gelatin prepared in H2O
