Biomod/2014/VCCRI/LabBook/Exp3
<html>
<link rel="stylesheet" href="http://fonts.googleapis.com/css?family=Lato:300,100&subset=latin"> <script src="//ajax.googleapis.com/ajax/libs/jquery/1.10.2/jquery.min.js" ></script>
<script type"text/javascript"> $(function () { $("style[media*='screen']").remove(); $("link[href*='favicon']").remove(); //fix heading var h1 = $(".firstHeading").text().split("/"); $(".firstHeading").text(h1[h1.length-1]); $("tr:odd").addClass("odd"); }); $('link[rel="shortcut icon"]').attr('href','http://openwetware.org/images/2/29/2014-EchiDNA-WEB-FAVICON.png'); </script> <link rel="icon" type="image/png" href="http://openwetware.org/images/2/29/2014-EchiDNA-WEB-FAVICON.png">
<style type="text/css"> /**** Base styles ****/ /*#column-one, */
- content {
font-weight: bold;
}
- footer, div#sidebar-main, #contentSub, .firstHeading, #siteSub, #jump-to-nav, .printfooter, #p-cactions {
display: none;
}
/*only show edit button - also remove p-cactions from previous style*/
- ca-nstab-main, #ca-talk, #ca-history, #ca-move, #ca-watch, #ca-link, #p-personal{
display: none;
}
- ca-edit{
display:block;
} /*Make text on edit buttons visible for easier editing*/ .editButtons, #wpSummary{ color: black; }
</style>
<meta charset="utf-8">
<meta http-equiv="X-UA-Compatible" content="IE=edge">
<meta name="viewport" content="width=device-width, initial-scale=1">
<title>EchiDNA 2014</title>
<link href="http://openwetware.org/index.php?title=Biomod/2014/VCCRI/bootstrapcss320&action=raw&ctype=text/css" rel="stylesheet">
<style type="text/css">
@font-face { font-family: BIOMOD_font; src: url(http://openwetware.org/images/9/9a/EchiDNA-2014-GothamRnd-Book.otf); }
@font-face { font-family: HEADING_font; src: url(http://openwetware.org/images/2/2f/EchiDNA-2014-KGSecondChancesSolid.ttf); }
@font-face { font-family: HEADING_ACTIVE_font; src: url(http://openwetware.org/images/2/2f/EchiDNA-2014-KGSecondChancesSolid.ttf); }
@font-face { font-family: FOOTER_font; src: url(http://openwetware.org/images/9/9a/EchiDNA-2014-GothamRnd-Book.otf); }
/*Alter default header*/
.navbar-default .navbar-nav > li > a { font-family: HEADING_font, Arial, sans-serif; }
.navbar-default .navbar-nav > li > ul > li > a {
font-family: HEADING_font, Arial, sans-serif;
font-size: 10pt;
}
.navbar-default .navbar-right > li { color: #f8f8f8; } .navbar-default .navbar-nav > .active > a, .navbar-default .navbar-nav > .active > a:hover, .navbar-default .navbar-nav > .active > a:focus { color: #F7941E; background-color: #F8F8F8; font-family: HEADING_ACTIVE_font, Arial, sans-serif; }
a[href^="mailto"] { color: white; }
a[href^="mailto"]:hover { color: #f7941e; text-decoration: none; }
a#vccri, a#edit-link { color:white; }
a#vccri:hover, a#edit-link:hover { color: #f7941e; text-decoration: none; }
body { background-image: url(http://openwetware.org/images/8/81/2014-EchiDNA-WEB-BG-TILE.png); background-repeat: repeat; color:white; font-family: BIOMOD_font, Arial, sans-serif; }
.col-centered { display:inline-block; float:none; text-align:left; /* inline-block space fix */ margin-right:-4px; }
h4 { color: #f7941e; font-family: HEADING_font, Arial, sans-serif; }
/*Footer stuff*/
html { position: relative; min-height: 100%; }
body { margin-bottom: 35px; }
.footer { position: absolute; bottom: 0; width: 100%; height: 35px; background-color: #0c2850; }
/*Fix OpenWetWare Editing Box*/
- wpTextbox1 {
color: #000; font-family: Arial; width: 100%; }
</style>
<html lang="en">
<head>
<script type="text/javascript"> $('#lab_book_link').addClass('active'); </script>
<style type="text/css">
- next-link {
color: #0F3264; text-decoration: none; margin-left: auto; margin-right: auto; font-size: 12pt; }
- next-link:hover {
color: #F7941E; }
.col-centered { display:inline-block; float:none; text-align:left; /* inline-block space fix */ margin-right:-4px; }
#LAB-BOOK-TITLE { font-family: HEADING_font, Arial, serif; color: black; font-size: 16pt; padding-top: 35px; }
h2 { font-family: HEADING_font, Arial, serif; color: #0F3264; font-size: 14pt; }
orange { color: #F7941E; }
#LAB-BOOK-TOP { display:block; width: 766px; height: 76px; margin-left: auto; margin-right: auto; margin-bottom: 0px; background-image: url(http://openwetware.org/images/9/9f/2014-EchiDNA-LAB-BOOK-BACKGROUND-TOP.png); }
#LAB-BOOK-REPEAT { display:block; position: relative; width: 766px; height: auto; margin-left: auto; margin-right: auto; margin-bottom: 0px; padding-bottom: 30px; padding-left: 60px; padding-right: 20px; background-image: url(http://openwetware.org/images/e/e9/2014-EchiDNA-LAB-BOOK-BACKGROUND-REPEAT.png); background-repeat: repeat-y; background-position: top; }
#LAB-BOOK-TEXT { position: relative; padding-top: 15px; color: black; text-align: justify; }
#LAB-BOOK-DIRTY-BOOK { display:block; width: 165px; height: 133px; background-image: url(http://openwetware.org/images/9/97/2014-EchiDNA-LAB-BOOK-EXPERIMENT-DIRTY-BOOK.gif); background-repeat: no-repeat; background-position: top;
position: absolute; top: 15px; left: 530px;
} #LAB-BOOK-DIRTY-BOOK:hover { background-position: bottom; }
#LAB-BOOK-CLEAN-BOOK { display:block; width: 167px; height: 129px; background-image: url(http://openwetware.org/images/4/4a/2014-EchiDNA-LAB-BOOK-EXPERIMENT-CLEAN-BOOK-LINK.png); background-repeat: no-repeat; background-position: top;
position: absolute; top: 20px; left: 110px;
} #LAB-BOOK-CLEAN-BOOK:hover { background-position: bottom; }
.image-left { font-size: 9pt; width: 50%; float: left; text-align: center; padding: 10px 15px; }
.image-right { font-size: 9pt; width: 50%; float: right; text-align: center; padding: 10px 15px; }
.image-center { font-size: 9pt; width: 100%; float: left; text-align: center; padding: 10px 15px; }
.image-right img, .image-left img { max-width: 91%; }
.image-center img { max-width: 95%; }
</style>
</head> <body>
<img src="http://openwetware.org/images/8/81/2014-EchiDNA-LAB-BOOK-EXPERIMENT-CLEAN-BOOK.png" /> <a href="http://openwetware.org/wiki/Biomod/2014/VCCRI/LabBook" id="LAB-BOOK-CLEAN-BOOK"></a> <a href="http://biomodaustralia2014.postach.io/" id="LAB-BOOK-DIRTY-BOOK" target="_blank"></a>
Aim
To generate single-stranded DNA of sufficient quantity and quality for assembling our cooperative biosensor.
Background
The “origami" in <a href="http://www.nature.com/news/2010/100310/full/464158a.html">DNA origami</a> describes the “folding" of long single-stranded DNA (ssDNA) scaffolds by hybridisation with short oligos, or "staples". Use of long scaffold strands which span the length of the entire structure appears to improve yields by reducing the possibility of small partial structures forming.
Fig 1. Scaffold strands "folding" into DNA origami by hybridisation with staples.
Traditionally, the scaffold strands used in DNA origami are actually the genomes of viruses! This is because viruses are the most diverse forms of life on Earth, and have found all kinds of crazy ways to store the information in their genome - for instance, in ssDNA. Many DNA origami have been made using the circular ssDNA genome of bacteriophage M13. However, our <a href="http://openwetware.org/wiki/Biomod/2014/VCCRI/LabBook/Coop">design</a> required a much smaller scaffold strand (912nt rather than around 7249nt).
Fig 2. Schematic of the barrel at the core of our cooperative biosensor, highlighting the scaffold in blue.
We researched and attempted three different methods of generating single-stranded DNA to build our cooperative biosensor:
- <orange>Asymmetric PCR: </orange> One primer is added in excess or exclusively over the other primer so than an ssDNA product is formed in each cycle. The concentration of the target ssDNA increases linearly over time.
- <orange>Biotin primer PCR & streptavidin beads:</orange> PCR using a biotinylated primer, resulting in a PCR product where one strand contains a 5' biotin group capable of binding to <a href="http://en.wikipedia.org/wiki/Streptavidin">streptavidin</a>. The dsDNA PCR product is denatured by NaOH or heat and the biotinylated strands removed using streptavidin beads.
- <orange>Phosphorylated primer PCR & lambda exonuclease:</orange> PCR with a phosphorylated primer, meaning that one strand of the PCR product contains a phosphate group preferentially digested by lambda exonuclease.
In preliminary trials, we found that we only produced detectable amounts of ssDNA using the third method, so we proceeded with this when producing sufficient ssDNA for further high-yield assembly of our cooperative biosensor. In the end, we had to use TWO techniques in series in order to obtain high enough yields of ssDNA for our experiments to structurally characterise the biosensor.
We used a phosphorylated PCR product as the template for an asymmetric PCR, and then used exonuclease to remove any leftover phosphorylated template while simultaneously increasing the yield of our scaffold strand.
Fig 3. Workflow for the generation of ssDNA scaffold for DNA origami experiments.
We were generating our ssDNA scaffold from scratch, so the sequence we used could be anything at all! Since the design of our cooperative biosensor was inspired by the switching mechanism of the <a href="http://openwetware.org/wiki/Biomod/2014/VCCRI/Project/Problem">bacterial flagella motor</a>, we decided to use the same DNA sequence responsible for the proteins in the flagella as the scaffold of our DNA origami.
Methods and Materials
PCR
This is a pretty standard procedure in molecular biology, so we’re going to assume you’ve learned about it elsewhere. The only trick we added was including primers that were chemically modified during DNA synthesis.
Fig 4. Table indicating PCR mastermix and thermocycle protocol.
Because we were conducting a super-scaled-up PCR (around 20ml total), but conducted the PCR in small volumes (around 100ul), we systematically sampled a few of the PCR reactions to confirm the purity and length of the DNA amplified during PCR.
Fig 5. Gel demonstrating correct amplification of FliG template.
Phenol/chloroform extraction and ethanol precipitation
This is also pretty standard, but you could learn more from <a href="http://cshprotocols.cshlp.org/content/2006/1/pdb.prot4455">Cold Spring Harbor</a> or from <a href="http://openwetware.org/wiki/Isopropanol_Precipitation_for_PCR_Purification">OpenWetWare</a>.
- Put 100% ethanol on ice.
- Combine PCR product into a single volume.
- Add an equal volume of phenol:chloroform:isoamyl.
- Mix until an emulsion forms and centrifuge at 12,000g for 10 minutes at room temperature. Organic and aqueous phases should be well-separated.
- Transfer aqueous phase to a fresh tube or container.
- Add an equal volume of chloroform:isoamyl.
- Mix until an emulsion forms and centrifuge at 12,000g for 10 minutes at room temperature. Organic and aqueous phases should be well-separated.
- Transfer aqueous phase to fresh tube (this hopefully contains your DNA).
- Estimate volume of your DNA solution.
- Add one-fifth volume of 3M sodium acetate.
- Add two volumes of ice-cold ethanol, allow DNA to precipitate on ice for at least half an hour (can be overnight).
- Centrifuge at 12,000g for 30 minutes at 0 degrees celcius (ought to be a fat, white pellet of DNA).
- Remove supernatent.
- Add one volume 70% ethanol, washing sides of tubes, and centrifuge at 12,000g for another 10 minutes at 0 degrees.
- Remove supernatent.
- Leave the open tube at room temperature until remaining ethanol completely evaporates.
- Redissolve pellet in 1ml of solvent (TE, EB, H20, etc.)
- Nanodrop to confirm concentration
Asymmetric PCR
The second round of PCR added the tricky bit - using a single primer in excess rather than two primers. This means that in each cycle of the PCR amplifies only a single strand - our DNA origami scaffold. Other slight alterations include a very high concentration of template DNA, an excess of nucleotides and a long extension step in each cycle of the PCR.
Fig 6. Table indicating asymmetric PCR mastermix and thermocycle protocol.
We optimised this reaction by running a time series to see how many cycles produced an optimal amount of our scaffold before the single strand began randomly self-priming and generating a range of longer amplicons, and also a concentration series to find the optimal concentration of template DNA.
Fig 7. Gel indicating time vs. concentration series for asymmetric PCR.
Exonuclease digestion
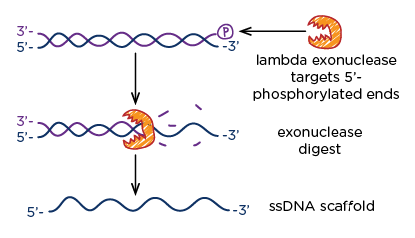
This step involved the use of lambda exonuclease - an enzyme that eats away at 5' ends of DNA, with a preference for phosphorylated ends. Since our initial dsDNA PCR product included a phosphorylated primer, we were able to remove leftover dsDNA from the asymmetric PCR reaction while simultaneously increasing the yield of our scaffold strand.
Fig 8. Exonuclease chews up the phosphorylated 5' ends of DNA.
We played with this part of the protocol a lot trying to <a href="http://link.springer.com/article/10.1007/s11274-010-0563-8"> optimise the yield of ssDNA</a>. For instance, we tuned the concentrations of enzyme:substrate DNA, the reaction times and even tried including a 5’biotin modification on the opposite strand to the 5’ phosphorylated base, in the hope that it would limit the degradation of our scaffold strand by the exonuclease.
We found that we could never completely remove the original dsDNA - perhaps because such a reaction would require an infinite amount of time, but perhaps also because of a limited yield of phosphorylation during synthesis of our primers.
However, we also found that we didn't needed to remove the DNA from the thermopol buffer, meaning that we could streamline our en masse PCR reaction straight to exonuclease digestion by just doing these three things:
- Add 11.5ul 10x lambda exonuclease buffer (NEB) to each 100ul PCR reaction.
- Followed by 5ul lambda exonuclease (NEB) to each 100ul PCR reaction.
- Then chuck the plate back in the thermocycler, for 35 minutes at 37 degrees celcius and 15 minutes at 75 degrees to heatkill the exonuclease.
Phenol/chloroform extraction and ethanol precipitation
We had to do this AGAIN!! This is because the polymerase and exonuclease needed to be removed. This step is also useful because we can resuspend the precipitated DNA in sterile filtered water, which allowed us to concentrate the ssDNA scaffold 100x to around 5ug/ul, ready for annealing with staples.
We concentrated our DNA using a ThermoScientific SpeedVac. Make sure that if you try this at home that you concentrate your DNA in pure water - any salts or buffering substances will obviously become more and more concentrated as well, potentially making your DNA more and more unconfortable!
Results
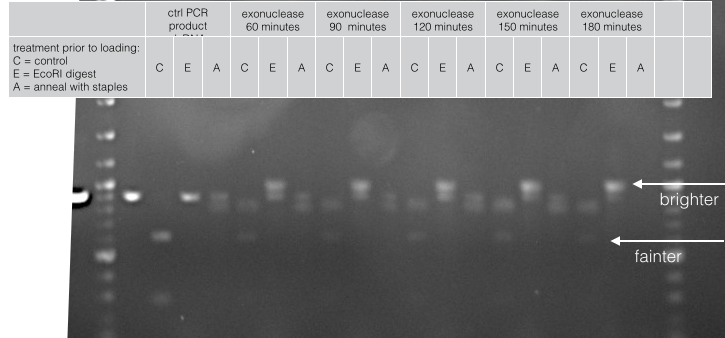
We used two experiments to characterise the different methods of generating ssDNA; digestion of dsDNA by restriction enzymes and assembly of the barrel at the core of our cooperative biosensor. This is because the original PCR product will cleave into two clean bands if digested with EcoRI and is unaffected by an annealing reaction with staples. In contrast, ssDNA is unaffected by EcoRI and hopefully doubles in molecular weight upon annealing with staples.
For instance, this figure shows how we optimised the time of exonuclease digestion. Notice how the dsDNA decreases over time while the annealed structure increases.
Fig 9. Exonuclease digestion of phosphorylated PCR product over time.
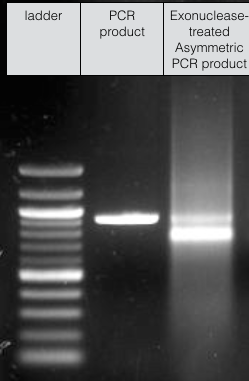
We were able to produce sufficient ssDNA for high-concentration DNA origami experiments, however, despite our best efforts our final scaffold solutions were still contaminated with some of the original dsDNA -
Fig 10. Scaffold for DNA origami experiments.
Conclusion
The generation of ssDNA was an essential step towards building our cooperative biosensor. In this set of experiments we found an optimal set of conditions for generating high-yields of ssDNA. This allowed us to assemble enough of our DNA origami for further structural and functional characterisation.
<a id="next-link" href="http://openwetware.org/wiki/Biomod/2014/VCCRI/LabBook">Click here to go back to the Lab Book Overview</a>
</body>
</html>
<html>
<script src="https://ajax.googleapis.com/ajax/libs/jquery/1.11.1/jquery.min.js"></script> <script src="http://openwetware.org/index.php?title=Biomod/2014/VCCRI/boostrap320&action=raw&type=text/javascript"></script>
<script type="text/javascript"> var pathArray = window.location.pathname.split( '/' ); var newPathname = ""; for (i = 2; i < pathArray.length; i++) { newPathname += pathArray[i]; newPathname += "/"; } newPathname=newPathname.slice(0,-1); document.getElementById('edit-link').setAttribute('href', 'http://openwetware.org/index.php?title='+newPathname+String.fromCharCode(38)+'action=edit'); </script>

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}