Biomod/2012/UTokyo/UT-Hongo/Assembly
<html> <head> <style>
- column-one { display:none; width:0px;}
.container{background-color: #ffffff; margin-top:0px} .OWWNBcpCurrentDateFilled {display: none;}
- content { width: 0px; margin: 0 auto auto 0; padding: 0em 0em 0em 0em; align: center;}
- column-content {width: 0px; float: left; margin: 0 0 0 0;padding: 0;}
.firstHeading {display:none; width:0px;}
- globalWrapper{width:984px; background-color: #ffffff; margin-left: auto; margin-right: auto}
- column-one {display:none; width:0px;background-color: #ffffff;}
- content{ margin: 0 0 0 0; align: center; padding: 12px; width: 960px;background-color: #ffff; border: 0;}
- bodyContent{ width: 960px; align: center; background-color: #ffffff;}
- column-content{width: 984px;background-color: #ffff;}
- footer{display: none; position: center; width: 960px}
@media screen {
body { background: #ffffff url(http://openwetware.org/images/1/14/Biomod-2012-utokyo-uthongo-wrapper-bg.jpg) repeat; }
}
</style>
<style>
- {
text-shadow: 0px 1px 2px #aaa; }
- bodyContent, #column-content, #content {
background: #ffffff url(http://openwetware.org/images/1/14/Biomod-2012-utokyo-uthongo-wrapper-bg.jpg) repeat; }
- column-content {
border-left: 1px solid #888; border-right: 1px solid #888;
}
- header {
width: 960px; height: 304px; background: url(http://openwetware.org/images/3/3b/Biomod-2012-UTokyo-UT-Hongo_header_test2.png) no-repeat; background-size: 100% auto; margin-bottom: 0px; padding: 0px 0px 0px 0px;
/* border-bottom: 1px solid #DEDEDE; */ }
- menu {
display: block; width: 956px; padding: 0px; background-color: black; border: 2px solid #888; margin-bottom: 15px;
}
ul.menu li a {
display: block;
/* border: 1px solid #474655; */
padding: 8px 10px; text-decoration: none;
/* color: #333; */
color: #fff;
/* width: 121px; */
margin: 0px; text-align: center; font-size: large;
}
ul.menu li a:hover{
color: cyan;
}
ul.menu { /* display: none;
position: absolute; */ height: 40px; margin:0; padding:0; list-style:none; float: center;
}
ul.menu li { /* display:inline;*/
margin:0; padding:6px; float: left; position: relative;
}
ul.menu li:hover ul {
display: block; position: absolute; z-index: 100;
}
ul.submenu {
display: none; list-style: none; background-color: #f0f0f0; border: 1px solid #ccc;
}
ul.submenu li {
float: none; margin: 0px;
}
ul.submenu li a {
text-align: left; margin: 0px; width: 340px; padding: 0 0 0 12px; color: black; text-decoration: none; font-size: large
}
ul.submenu li a:hover {
color: black; text-decoration: underline;
}
.thumb {
border-image: url(http://openwetware.org/images/1/14/Biomod-2012-utokyo-uthongo-wrapper-bg.jpg) repeat;
}
.mytable {
border-image: url(http://openwetware.org/images/1/14/Biomod-2012-utokyo-uthongo-wrapper-bg.jpg) repeat;
}
</style> </head> <body>
<a href="http://biomod.net/"><img src="http://openwetware.org/images/8/82/Biomod2012-logo.png" width="250px" height="50px"></img></a>
<a href="http://www.u-tokyo.ac.jp/en/" target="_blank"><img src="http://www.u-tokyo.ac.jp/en/images/banner/UT-logo.gif" width="234" height="60" border="0" alt="The University of Tokyo"></img></a>
</body> </html>
<html>
<style>
ul.menu li.</html>result<html> a {
color: cyan;
}
- toc {
display: none;
}
- mytoc {
background: #dedede; width: 200px;
}
- toc ul ul, .toc ul ul {
margin: 0 0 0 1em;
}
- mytoc a, #mytoc a:visited {
font-size: normal; color: black;
}
- mytoc a:hover {
font-weight: bold; text-decoration: none;
}
- wiki-toc {
width: 200px; margin-top: 6px; float: left;
}
- wiki-body {
margin-left: 200px; padding-left: 12px;
}
- toctitle span {
display: none;
}
- wiki-body p,
- wiki-body li,
- wiki-body dd,
div.thumbcaption {
font-size: medium;
}
</style>
<script type="text/javascript">
window.onload = function () {
document.getElementById('mytoc').appendChild(document.getElementById('toc').firstChild);
};
</script>
<html>
<html>
<a href="http://openwetware.org/index.php?title=Biomod/2012/UTokyo/UT-Hongo/</html>Assembly<html>&action=edit">edit this page</a>
<html><style> ul.menu li.result ul.submenu li a {
color: black;
}
ul.menu li.result ul.submenu li a:hover {
text-decoration: underline;
} </style></html>
Design of the DNA Sequence
In this section, we will explain the design of our DNA Shell in detail. The DNA Shell is constructed of DNA Origami. In this section, we explain about DNA Origami, and then describe about our design of the typical structure and added functions of DNA Shell. Also, we will describe the simulation techniques to calculate the melting temperature and the Gibbs free energy in hybridization.
DNA origami
DNA Origami is a complex 2D or 3D structure which is fabricated using helical structures of DNA. Generally, DNA Origami is composed of one scaffold DNA, which is a long single-stranded DNA, and many staple DNAs, which are short single-stranded DNAs. Staple DNAs are designed so that they connect the parts of the scaffold DNA to make the desired structure. In designing DNA Origami, the phase of hydrogen bonding of DNA helical structure should also be concerned. We designed the DNA sequence of the DNA Shell using CADNano®, the software for designing DNA Origami.
Main Structure of DNA shell
As materials of DNA shell, we used M13 as scaffold DNA, which is a typical material of DNA Origami. M13 has approximately 7250 base pairs and it is an ideal number for our shell. However, some parts of this DNA hybridizes by itself, therefore we had to cut those parts off. In designing the staple DNAs, we standardized the number of base pairs to 32 bp. This is important because the conditions of hybridization is greatly affected by the number of base pairs.

Fig. 1.1 is the picture of DNA Shell on CADNano®. The blue line is the scaffold M13 and the others are the staples. The shell has three large domains which are about 40nm × 30nm. The domains in the center and the right are the body of the shell. Various functional DNAs like the ones used for bonding to streptavidin could be attached. The left domain is used for attaching the DNA Shell on to a solid surface. In order to make the links between the domains loose enough so that the structure can fold, the staples to reinforce these areas were not added.
Adding Functionality


In order to add functionality to the body, we extended some of the staples so they could bond with single-stranded DNAs that are modified with some molecules (Fig. 1.2). Therefore, for the functional DNAs, we added 13 more base pairs to the staples which originally had 32 base pairs. We designed all these staples to extend from the same side of shell surface taking care of the phase of the hydrogen bond.
We needed to attach 12 single-stranded DNAs with fluorescent molecules (named as fluorescent DNAs) and two functional single-stranded DNAs to one side, and 12 single-stranded DNAs with quenching molecules (named as quenching DNAs) and two functional single-stranded DNAs to the other side. We had to think up of 4 different sequences that would bind to either:
- Fluorescent DNAs
- Quenching DNAs
- Functional DNAs to the fluorescent side
- Functional DNAs to the quenching side
We designed the structure so that the functional part is surrounded by fluorescent/quenching molecules (Fig. 1.3). To quench the light emitted by the fluorescent molecules when the shell is closed, the two molecules must be close to each other. Therefore, we designed the fluorescent molecules and the quenching molecules to face each other when the shell is closed.
Simulation Techniques
Simulation techniques were applied to make sure that the staples are hybridized only at the points where we want hybridization to occur. That is, we have to keep the energy for hybridization high in inappropriate areas and keep it low in the correct areas. In order to achieve this requirement, we focused on these three points.
- Make sure that the complementary arrangements of the designed staples does not appear in inappropriate area.
- Keep the melting temperature of correct hybridization low, and keep the melting temperature of incorrect hybridization high.
- Avoid steric hindrance.
The Watson-Crick base pair is stable. Therefore if there are no other possibilities of hybridization of the staples and the scaffold in the inappropriate areas, the Gibbs energy of the structure would become the lowest when hybridization occurs in the appropriate areas. This way, our first focus, hybridization at correct areas, will be achieved. Also, we accomplished the second requirement through simulation which is described below. If we hybridized the sequence extended from the staples and the functionalized DNAs at the same time, there would be some steric hindrance. To weaken this effect, we set a realm where the staples will not be hybridized. What we actually did was to embed two T (thymine) bases in the staples.
To simulate the energy of hybridization and melting temperature, we used the software named DINAMelt and NUPACK supported online. Since there is no absolute solutions for design, we needed to get over these difficulties by trial and error; It was serious and troublesome work. In designing the arrangement extended from staples, we need to attach the same arrangement to the staples attached with functional parts. This work of discovering moderate arrangement cost us immense labor, because it is required to make these staples added only ONE pattern of arrangement hybridized stably.

Fig. 1.4 is one example of simulation using DINAMelt (http://mfold.rna.albany.edu). This is a simulation to estimate the melting temperature and the possibility of correct hybridization. This is simulation was done under the situation of
- From 0°C by 1°C to 100°C as DNA
- [A0] = 1.6e-07 M, [B0] = 1.6e-07 M ;[A0] is concentration of extended staple and [B0] is concentration of modified functional staples
- [Na+] = 0.2 M, [Mg++] = 0.0125 M
The results of the simulation shows that the hybridization of A and B (which means the appropriate hybridization) is dominant at low temperature(from 0°C to 30°C). The data gained (melting temperature) was used for annealing process in the experiment.
Experimental Results
In this section, we write about the results to our experiment. Our experiment consists of four themes;
- Assembly of the DNA Shell
- Capturing Ability
- Immobilizing on Microfluidic Device
- Supporting Enzyme
Experiments to show the detection ability of the DNA Shell is included in the "Capturing Ability". The results are listed in order below.
Assembly of the DNA Shell
First, we mixed M13 and staples, and ascertained whether DNA Shell was hybridized as we had designed by agarose gel electrophoresis and atomic force microscope (AFM).
The staple DNA was mixed with M13 (160 or 16 nM of each staple DNA, 1.6 or 4 nM of M13, a 100 or 4 –fold excess of staple DNA) in 1x TAE/Mg buffer. When we added functional staples which are DNA with molecules such as the fluorescence, the quencher and the biotin instead of normal staples, we made the density of functional staples all the same as normal staples. Samples were annealed two hours from 95℃ to 20℃ in a thermal cycler at a rate of 6.25℃/10minutes 12 steps.
Agarose Gel Electrophoresis

We confirmed that we had the structure which had the bigger molecular weight than original M13(scaffold) by electrophoresis. Samples were electrophoresed in a 0.6 % agarose gel containing 1X TAE/Mg buffer. The agarose gel was run at 4℃ for 70 minutes (Fig.1 & 2).
The band of M13+staple ran longer than the band of M13. It showed that the structure which had big molecular weight was created. The lower fuzzy band was the band of excess staple DNA. When we put staples both 100 and 4 times of M13mp18, the annealing went well.
AFM

These DNA origami (M13 + all staples) were observed by using AFM to confirm that these DNA Shell were formed correctly. 1xTAE/Mg solution was utilized as buffer.
This AFM picture clearly shows that the origami with all staples was formed as designed. There was the point where it seemed that origami connected with each other aside. This is probably because each DNA Shell was attracted each other by π-π stacking interaction. We designed DNA Shell as base pair lined lengthways, so it was appropriate for DNA Shell to connect sideways by π-π stacking interaction.
Capturing Ability
Next, we ascertained whether DNA Shell captured target molecules (target DNA, streptavidin) by using agarose gel electrophoresis, fluorometry, and AFM.
Agarose Gel Electrophoresis

We ascertained whether the migration distance changed after target DNA was added in the solution with DNA Shell. After having annealed the sample which added target DNA (control1 and control2) to M13 and staple, we electrophoresed and confirmed a change of the phoresis distance. Samples were electrophoresed in a 0.6 % agarose gel containing 1X TAE/Mg buffer at 4℃ for 70 minutes.
All the samples which were added target DNA to come to have a shorter phoresis distance than the sample which wasn’t add. It is thought that it became hard to go through the mesh of the gel by Shell having been closed.
Fluorometry
After having annealed the sample which contained the scaffold (M13), staple DNA, DNA with a fluorescence molecule and DNA with quencher molecule, we took 100μl of the sample and added 400μl of 1xTAE/Mg buffer and measured the fluorescence intensity. We recorded the fluorescence change before and after we put the target DNA and streptavidin (SA). The target DNA is a DNA fragment that is made to close the DNA Shell instead of streptavidin.
Fluorescence Change (1 equiv. of target DNA)
Step 1

After having added target DNA, intensity of fluorescence decreased. It is thought that the fluorescence decreased because Shell closed by having put target DNA and fluorescence molecules approached quencher molecules.
Step 2

The next graph is the change of the peak numerical value when we adjusted the density of target DNA. We calculated numerical value after having added target DNA for 100 at the intensity of fluorescence of the peak before adding target DNA.
Step 3

We changed the number of fluorescence molecules and the quencher molecules which spread from the Shell surface (12, 4 or 1) and, in the case of each, checked how became a fluorescence change. In the lower graph, peak numerical value before adding target DNA is 100.
-
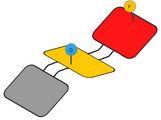 DNA Shell with one pair of Fluorescent and Quencher molecules
DNA Shell with one pair of Fluorescent and Quencher molecules -
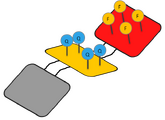 DNA Shell with four pairs of Fluorescent and Quencher molecules
DNA Shell with four pairs of Fluorescent and Quencher molecules -
 DNA Shell with twelve pairs of Fluorescent and Quencher molecules
DNA Shell with twelve pairs of Fluorescent and Quencher molecules



Step 4

In the case of four and twelve pairs of fluorescent and quencher molecules there is not big difference, but there is a significant difference if we compare with the case of just one pair of fluorescent and quencher molecule. It can be said that the detection ability would increase when multiple pairs of fluorescence and quencher molecules are attached.
Following target DNA, we checked whether we could capture streptavidin with DNA Shell.
After having annealed the sample which contained M13, staple DNA, DNA with a fluorescence molecule, DNA with quencher molecule and DNA with biotin, we took 100μl of the sample and added 400μl of 1xTAE/Mg buffer and measured the fluorescence intensity. We recorded the change in fluorescence before and after we put SA.
Fluorescence Change (1 equiv. SA)
Adding SA

After having added SA, intensity of fluorescence decreased. It is thought that the fluorescence decreased because the Shell closed and the distance between fluorescence and quencher molecules became small causing quenching of the light.
Changing the Concentration of SA

The next graph shows the change in the peak fluorescence when we changed the concentration of SA. We calculated using the peak value after normalizing the fluorescence before adding SA as 100.
The biggest change in fluorescence was observed when 1/8 equiv. of SA was added. It was thought that the change in fluorescence become small when SA concentration was high, because two molecules of SA would connect to one DNA Shell and prevent the DNA Shell from closing.
-
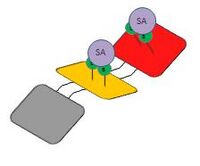 In case of high concentration
In case of high concentration -
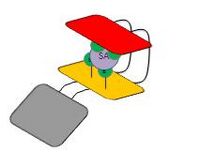 In case of low concentration
In case of low concentration -
 In case of absence of SA
In case of absence of SA



Fluorometry
We performed fluorometry in three kinds of Shell ([1] Shell where biotin grew on both sides (Normal Shell) [2] Shell where biotin grew only in the fluorescence side [3] Shell where biotin grew only in the quencher side).
-
![[1] Shell where biotin grew on both sides (Normal Shell)](https://oww-files-thumb.sfo3.cdn.digitaloceanspaces.com/1/16/Biomod-2012-UToky%E2%80%8Bo-UT-Hongo-bothsides.png/200px-Biomod-2012-UToky%E2%80%8Bo-UT-Hongo-bothsides.png) [1] Shell where biotin grew on both sides (Normal Shell)
[1] Shell where biotin grew on both sides (Normal Shell) -
![[2] Shell where biotin grew only in the fluorescence side](https://oww-files-thumb.sfo3.cdn.digitaloceanspaces.com/1/11/Biomod-2012-UToky%E2%80%8Bo-UT-Hongo-fluorescenceside.png/200px-Biomod-2012-UToky%E2%80%8Bo-UT-Hongo-fluorescenceside.png) [2] Shell where biotin grew only in the fluorescence side
[2] Shell where biotin grew only in the fluorescence side -
![[3] Shell where biotin grew only in the quencher side](https://oww-files-thumb.sfo3.cdn.digitaloceanspaces.com/1/1c/Biomod-2012-UToky%E2%80%8Bo-UT-Hongo-quencherside.png/200px-Biomod-2012-UToky%E2%80%8Bo-UT-Hongo-quencherside.png) [3] Shell where biotin grew only in the quencher side
[3] Shell where biotin grew only in the quencher side
![[1] Shell where biotin grew on both sides (Normal Shell)](/wiki/File:Biomod-2012-UToky%E2%80%8Bo-UT-Hongo-bothsides.png)
![[2] Shell where biotin grew only in the fluorescence side](/wiki/File:Biomod-2012-UToky%E2%80%8Bo-UT-Hongo-fluorescenceside.png)
![[3] Shell where biotin grew only in the quencher side](/wiki/File:Biomod-2012-UToky%E2%80%8Bo-UT-Hongo-quencherside.png)


There was no big difference between [3] and [1]. However, only [2] came to have a particularly big fluorescence change.
Two molecules of biotin is growing from one domain of the Shell in [2] and [3]. Because four molecules of biotin can bind with one molecule of SA, it is thought that two Shell catched one SA, and a dimer was formed. In this case a large number of fluorescence molecules would approach at a time in [2], and it is thought that quenching occured and the fluorescence decreased.

Fig. 2.2.3.3.3 Dimer
AFM

First, we observed the DNA Shell that captured the target DNA. This AFM picture shows that the target DNA was captured. In the picture, some square shapes could be seen. They are the DNA Shell.
We found that the thickness of the white point in the Shell (Fig.2.2.3.1) was about 4.9nm(Fig. 2.2.3.2). In this picture, the left upper graph shows the sectional pitch that cut Shell perpendicularly. Target DNA is 26 bp or about 5.8 nm, so we estimated that the target DNA hybridized with staple DNA with a little bending.
-
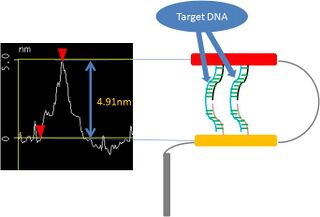 Fig.2.2.3.2 Analysis image of closing DNA Shell
Fig.2.2.3.2 Analysis image of closing DNA Shell -
 Fig. 2.2.3.3 Closing DNA Shell with Streptavidin
Fig. 2.2.3.3 Closing DNA Shell with Streptavidin


Following target DNA, we checked whether we could see the DNA Shell that had captured streptavidin. At the upper left of the above picture, we could see a square structure which has two white points in the center. This structure is the Shell. The following pictures show the sectional pitch.




By analyzing the AFM pictures we could see that:
- Shell is 91.797 nm (almost 90 nm) wide
- Shell is 2.075 nm (almost 2nm) thick
- The white points of Shell are 3.882 nm and 4.096 nm(almost 4 nm) thick
- There is a part with almost 0 nm thick at around 21 nm from the side of Shell
Shell is constructed from bundles of double helix, so it is reasonable enough to think that the value 2 nm is Shell's thickness. (The diameter of the double helix is 2 nm). The thickness at the white points in the Shell are equivalent to the thickness for approximately two folds of double helix. In addition, width of Shell at the design stage is approximately 120 nm and the structure seen this time, width is almost 2/3 of it. It is reasonable to suppose that this is because one origami domain is on top of another domain. From this geometrical information, we could conclude that the Shell had successfully captured streptavidin. The white points of the Shell are smaller compared with the Shell itself because streptavidin is small (almost 60 Å)
<html>Immobilizing on Microfluidic Device
We confirmed DNA origami is immobilized on the surface of microfluidic device. For the adsorption of DNA origami to clean or hydrophilic silicon dioxide surfaces, glass was cleaned with acetone and 2-propanol, then rinsed in DI water, assisted by ultrasonic agitation. Next, the surface was treated with oxygen plasma to yield a hydrophilic surface, using standard cleaning parameters (50% power and 0.5 mbar chamber pressure for 30 sec). A drop of 2 μl 5× TAE buffer was dispensed onto the cleaned surface. The TAE buffer spreaded completely over the cleaned surface. A 2 μl drop of DNA origami solution was dispensed on the top. The incubation time was 30 minutes in a closed petri dish. After incubation the sample was dipped for 5 seconds into a solution of water and ethanol (50:50 v/v), followed by immersion for one hour in a solution of water in ethanol (10:90 v/v). All steps were done at room temperature.
Target DNA was introduced into Microfluidic Device
_ver5.jpg)
- We made 3 microfluidic devices for comparing experiment. We introduced 4μl of solution containing fluorescent labeled-DNA origami(M13 + staples) into every microfluidic devices. After immobilizing DNA origami, we rinsed microchannel 3 times by TAE Buffer to remove DNA origami which is not immobilized on the microfluidic. After rinsing, we kept one device for reference data, introduced water into another device, and control solution into the other device. After that, observation of fluorescence from immobilized DNA origami was performed
- The fluorescence of reference microchannel (a). The scale bar is 100 μm.
- The fluorescence was almost same even if water is introduced into the microchannel (b).
- The fluorescence strength decreased when the control solution is introduced into the microchannel (c).
And we showed the fluorescence intensity of each image to the graph.(Fig. 2.3.1.1)
_ver6.jpg)
After water was introduced, the decrease in the fluorescence intensity was observed due to destruction of immobilized DNA origami. It is caused by salt concentration change. Also after target DNA was introduced, fluorescence intensity decreased greatly. This shows that the target DNA closed the DNA Shell.
Therefore, we confirm that DNA origami was immobilized onto the surface of the microchannel and works as we expected.
Introduction of Streptavidin into Microfluidic Device

- We made 3 microfluidic devices for comparing experiment. We introduced 4μl of solution containing fluorescent labeled-DNA origami(Biotin attached) into every microfluidic devices. After immobilizing DNA origami, we rinsed microchannel 3 times by TAE Buffer to remove DNA origami which is not immobilized on the microfluidic. After rinsing, we kept one device for reference data, introduced water into another device, and streptavidin into the other device. After that, observation of fluorescence from immobilized DNA origami was performed
- The fluorescence of reference microchannel (d). The scale bar is 100 μm.
- The fluorescence was almost same even if water is introduced into the microchannel (e).
- The fluorescence strength decreased when streptavidin is introduced into the microchannel (f).
And we showed the fluorescence intensity of each image to the graph.(Fig. 2.3.2.1)
_ver4.jpg)
Biotin attached labeled-DNA origami isn't immobilized on microchannel and isn't rinsed completely. So after water was introduced, fluorescence intensity greatly fell. Also fluorescence intensity became weak after the streptavidin was introduced. Fluorescence decreased because the streptavidin was attached to the DNA origami immobilized on the microchannel.
<html>Supporting Enzyme
Based on the results mentioned above, we did further advanced experiments. We confirmed whether DNA Shell supported enzymes.
Background
We used 3,5,3’,5’-tetramethylbenzidine (TMB), streptavidin with horseradish peroxidase (HRP) labeling and trypsin. TMB can be oxidized for the reduction of hydrogen peroxide (H2O2) to water by such as HRP.
We make use of two biochemical reactions;

(1) TMB is oxidized by H2O2/HRP. This reaction happens with a change of color. When TMB (λmax=285nm) is oxidized, the solution takes on a blue color (λ=370 and 652nm) at first and gradually turns to yellow (λmax=450nm). ( Josephy, P. D., Eling, T., and Mason, R. P.(1982) J.Biol.Chem, 257, 3669-3675)
(2) Trypsin, a protease that cleaves peptide chains mainly, decomposes HRP. (Welinder, K. G. (1979) Eur. J. Biochem, 96, 483-502)

If trypsin decomposes HRP, before the reaction happens between TMB and HRP, HRP does not oxidize TMB. Furthermore, we used streptavidin-labeld HRP and DNA Shell with biotin. DNA Shell protects HRP because of the combination between streptavidin and biotin. As HRP protected by Shell, trypsin does not decompose HRP and the oxidization of TMB goes on.
Experiment Condition
We made four kinds of solution like below (Table 2.4.2) and measured the absorption spectra of each solution.
| TMB | HRP | Trypsin | DNA Shell | Expected Peak Wavelength[nm] | |
|---|---|---|---|---|---|
| No.1 | + | - | - | - | 285 |
| No.2 | + | + | - | - | 450 |
| No.3 | + | + | + | - | 285 |
| No.4 | + | + | + | + | 450 |
Table 2.5.2 Four kinds of solution for the experiments.
- (No.1) We only used 2μM TMB in acetate buffer.
- (No.2) We added 20nM streptavidin-labeled HRP in tris HCl to the solution of 2μM TMB in acetate buffer.
- (No.3) We added the solution in which we reacted trypsin and HRP for 1 hour in 37C constant temperature bath and HRP to TMB. As a whole solution, 0.2µM TMB, 2µMHRP, excess trypsin and 50mM acetate (TMB buffer), 50mM trisHCl (HRP buffer) are contained.
- (No.4) We added the solution in which we reacted trypsin, HRP and Shell for 1 hour in 37C constant temperature bath and HRP to TMB. 0.2µM TMB, 2µMHRP, 2nM DNA Shell, excess trypsin and 50mM acetate (TMB buffer), 50mM trisHCl (HRP buffer) are contained.
Results and Measurements
-
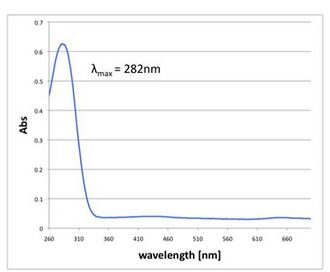 Fig 2.4.3.1 TMB Only. Extinction wavelength was peaked at 282nm, which is almost the same peak of TMB.
Fig 2.4.3.1 TMB Only. Extinction wavelength was peaked at 282nm, which is almost the same peak of TMB. -
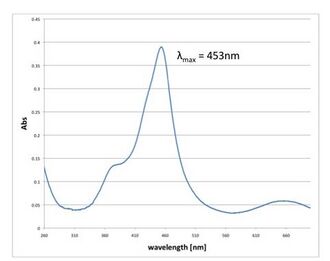 Fig 2.4.3.2 TMB and HRP-labeled streptavidin. Extinction wavelength was peaked at 453nm, which is almost the same peak of oxidized TMB.
Fig 2.4.3.2 TMB and HRP-labeled streptavidin. Extinction wavelength was peaked at 453nm, which is almost the same peak of oxidized TMB. -
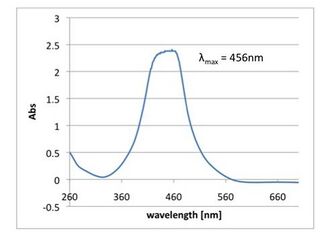 Fig 2.4.3.3 TMB, HRP-labeled streptavidin and trypsin
Fig 2.4.3.3 TMB, HRP-labeled streptavidin and trypsin -
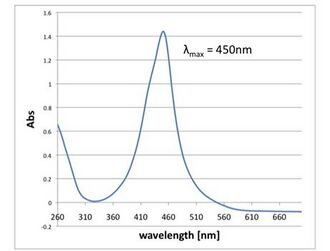 Fig 2.4.3.4 TMB, HRP-labeled streptavidin, trypsin and DNA Shell. The peak was 450nm, which is the same of oxidized TMB. We expected that DNA Shell protected HRP-labeled streptavidin.
Fig 2.4.3.4 TMB, HRP-labeled streptavidin, trypsin and DNA Shell. The peak was 450nm, which is the same of oxidized TMB. We expected that DNA Shell protected HRP-labeled streptavidin.




Lab Book
Please refer to below link.
<html></html>
<html> <head>
<style>
- myfooter {
border-top: 1px solid #a9a9a9; padding-top: 60px; margin-top: 18px;
}
- sitemap {
width: 934px; height: 250px; background: #f0f0f0; padding: 12px; border: 1px solid #808080;
}
- sitemap a {
color: #666;
}
- sitemap h3 a {
text-decoration: underline;
}
- sitemap a:hover {
}
- footer-contents {
padding: 12px; height: 140px;
}
- sitemap h2 {
color: #888;
}
.footer-section { width: 96px; float: left; margin-right: 30px; }
- section1 {
width: 64px; }
- section3 {
width: 124px;
}
- section4 {
width: 108px;
}
- section5 {
width: 135px;
}
- section6 {
width: 72px;
}
- section7 {
float: right;
}
.footer-section h3 {
font-size: small;
}
.footer-section ul li a {
font-size: small;
}
- copyright {
text-align: center; color: #aaa;
}
</style>
</head> <body>
<a href="http://openwetware.org/index.php?title=Template:Biomod/2012/UTokyo/UT-Hongo/Footer&action=edit">edit this footer</a>
</body> </html>

{kind=link}
{kind=link}
{kind=link}
{kind=link}