Biomod/2012/TU Dresden/Nanosaurs/Lab book
<html> <script src="http://biomod-dresden-2012.googlecode.com/svn/trunk/js/jquery-1.8.2.min.js"></script> <script src="http://biomod-dresden-2012.googlecode.com/svn/trunk/js/jquery-ui-1.9.1.custom.min.js"></script> <script src="http://biomod-dresden-2012.googlecode.com/svn/trunk/js/jquery.smooth-scroll.min.js"></script> <script src="http://biomod-dresden-2012.googlecode.com/svn/trunk/js/lightbox.js"></script> <script type="text/javascript" src="http://biomod-dresden-2012.googlecode.com/svn/trunk/js/jquery.queryloader2.js"></script> <script type="text/javascript" src="http://biomod-dresden-2012.googlecode.com/svn/trunk/js/superfish.js"></script> <script type="text/javascript" src="http://biomod-dresden-2012.googlecode.com/svn/trunk/js/jquery.hoverIntent.minified.js"></script> <script type="text/javascript" src="http://biomod-dresden-2012.googlecode.com/svn/trunk/js/jquery.scrollTo-1.4.3.1-min.js"></script> <script type="text/javascript" src="http://biomod-dresden-2012.googlecode.com/svn/trunk/js/jquery.localscroll-1.2.7-min.js"></script> <link href='http://fonts.googleapis.com/css?family=Merriweather:400,700' rel='stylesheet' type='text/css'> <link href='http://biomod-dresden-2012.googlecode.com/svn/trunk/css/superfish.css' rel='stylesheet' type='text/css'> <link rel="stylesheet" href="http://biomod-dresden-2012.googlecode.com/svn/trunk/css/lightbox.css" type="text/css" media="screen" /> <link rel="stylesheet" href="http://code.jquery.com/ui/1.9.0/themes/base/jquery-ui.css" /> <script type"text/javascript"> // Main function that waits for the browser to be ready $(document).ready(function(){ //make css accesible, please change the alter_css to chnage the style var alter_css = $("#alter_css").html(); $("style").remove(); $('head').append('<link rel="stylesheet" href="/skins/monobook/shared.css?164" type="text/css" />'); $('head').append('<link rel="stylesheet" href="http://biomod-dresden-2012.googlecode.com/svn/trunk/nanos.css" type="text/css" />'); //additional divs
$(".firstHeading").wrap('
'); $(".firstHeading").wrap('
'); $(".firstHeading").wrap('
');
var nav = $("#nav").html(); $('#inner_header').append(nav);
$('#inner_header').append('
');
//clean up wiki framework $("#sidebar-main").remove(); $(".portlet").remove(); //fix breadcrumbs $('#contentSub').remove(); //fix heading var h1 = $(".firstHeading").text().split("/"); $(".firstHeading").text(h1[h1.length-1]); //start plugins for navigation $("ul.sf-menu").superfish({ delay: 800, // one second delay on mouseout animation: {opacity:'show',height:'show'}, // fade-in and slide-down animation speed: 'normal', // faster animation speed }); $('#main_saurs').localScroll(); $("tr:odd").addClass("odd");
}); </script>
<script type="text/javascript"> var _gaq = _gaq || []; _gaq.push(['_setAccount', 'UA-35720700-1']); _gaq.push(['_trackPageview']);
(function() { var ga = document.createElement('script'); ga.type = 'text/javascript'; ga.async = true; ga.src = ('https:' == document.location.protocol ? 'https://ssl' : 'http://www') + '.google-analytics.com/ga.js'; var s = document.getElementsByTagName('script')[0]; s.parentNode.insertBefore(ga, s); })(); </script> <script type="text/javascript">var addthis_config = {"data_track_addressbar":false};</script> <script type="text/javascript" src="http://s7.addthis.com/js/300/addthis_widget.js#pubid=ra-508414b242f27ceb"></script> <script id="nav">
</script>
</html> <html> <script type="text/javascript" src="http://biomod-dresden-2012.googlecode.com/svn/trunk/js/jquery.tweet.js"></script> <script type"text/javascript"> $(document).ready(function(){ window.scrollTo(0, 155); $(".tweet").tweet({ count: 6, query: "from:nanosaurs", loading_text: "searching twitter..." });
});
$(document).ready(function(){ $("tr:odd").addClass("odd"); });
</script>
<body>
<a href="#recipes">Recipes</a> <a href="#protocols">Protocols</a> <a href="#downloads">Downloads</a>
<a id="recipes">
Recipes
DNA origami recipes
| Description | Folding Buffer 8mM MgCl2: Buffer to be added before DNA origami assembly to create the necessary surrounding conditions for correct folding and assembly. | ||
| Recipe | 1x | 10x | |
| 1M MgCl2 | 8µl | 80µl | |
| 1M Tris | 5µl | 50µl | |
| 0,5M EDTA | 2µl | 20µl | |
| H2O | 985µl | 850µl | |
| 1ml | 1ml | ||
| Description | Combines staple strands and scaffold in the necessary surrounding conditions. Staple strands have an excess of 7,5x in comparison to the scaffold to increase the chances of correct assembly. | |
| Recipe | 100nM p7560 single strand | 10µl |
| FB 8 10x | 10µl | |
| Staple mix (≈500nM per oligo) | 14,8µl | |
| H2O | 65,2µl | |
| 100µl | ||
| Description | The certain mix of different staples (oligonucleotides) that are needed to form different structures with their respective functions. | ||||
| Recipe | Staple type | Open | Closed guided | Closed not guided | |
| core | x | x | x | ||
| core_guideadj | x | x | |||
| guide_adj | x | ||||
| guide | x | ||||
| apt_locks | x | x | |||
| anchor | x | x | x | ||
| catcher | x | x | x | ||
| locks_nohang | x | If only the part inside the DNA origami and no overhang of the locks is wanted. | |||
| anchor_nohang | If anchor strands are not wanted. | ||||
| catcher_nohang | If catcher strands are not wanted. | ||||
| edge_stab_45 | For labeling of the DNA origami. | ||||
</body>
GUV recipes
| Description | SLB (supported lipid bilayer) buffer is used as the solution for all the vesicle experiments. |
| Recipe | 10mM HEPES |
| 150mM NaCl | |
| 8mM Mg2++ | |
| Recipe | Polyacrylamid | 3ml |
| H2O | 5,894ml | |
| TBE 10x | 1ml | |
| APS | 100µl | |
| TEMED | 6µl | |
| TdT Buffer | 12 | |
| CoCl2 | 12 | |
| Oligonucleotide | 100 µM | 3 |
| Alexa-dUTP conjugate/ Biotin-ddUTP conjugate |
1mM | 3 |
| Terminal Transferase | 3 | |
| Water | 27 | |
| Total | 60 | |
| Procedure | 1. Incubate at 370 C for 60 mins. | |
| 2. Heat inactivate at 700 C for 15 mins. | ||
| 3. Cleanup with NucleoSpinExtract II kit. | ||
| 4. Measure the resultant concentration using <a href="http://www.nanodrop.com/?gclid=CPT5qOzCorMCFYq-zAodFA4Axw">NanoDrop</a> | ||
Protocols
Giant unilamellar vesicles (GUVs) formation
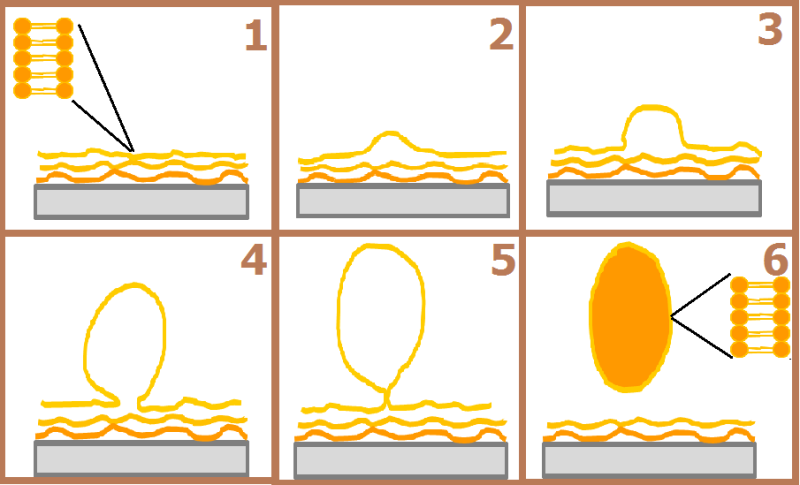
The method used to form the GUVs is “Vesicle Formation from emulsion”. This method consists in a controlled hydration of dried or nearly dried films of lipids deposited on a solid surface, in this case electrodes.
- Preparation of lipids solution
- Lipids spreading
- Solvent evaporation
- Electroformation
The lipid mixture contains 1, 2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1, 2-dioleoyl-sn-glycero-3-phosphoserine (DOPS) in a 9:1 proportion in 1 mg/ml concentration. The solution is prepared with the organic solvent chloroform.
6.5 µl of the lipid mixture is evenly spread in the surface of two little wires, the electrodes, which are previously cleaned by sonication cycles, washed with ethanol and dried.
The lipid sample is left to dry over the electrodes inside an extractor for at least 15 min to evacuate the solvent of chloroform.
The electrodes are inserted in the lid of a plastic chamber that is filled with 3.5 ml of sucrose solution isomolar to the working buffer (HEPES, NaCl and w/o Mg2+). This solution hydrates the dry lipid sample to form the GUVs emulsion. The hydration is carried out in the presence of an electric field with a frequency of 10 Hz during 2 hours to electroformate the vesicles and of 2 Hz during 30 min to detached them from the electrodes.
Large unilamellar vesicles (LUVs) formation
The method employed consists of rehydratation and extrusion, whose detailed procedures are summarized as following:
- Preparation of lipids solution
- Evaporation
- Hydratation
- Incubation
- Freeze/thaw
- Extrusion
- The Teflon piece is wetted with buffer.
- The 2 pieces of cellulose membrane are placed in a support in the center of the Teflon piece.
- Put the PC membrane with a pore size of 50-100 µm on the taller piece of the holder.
- A drop of buffer is placed on top of the membrane.
- The 2 pieces of Teflon holder are placed together.
- The syringe is filled with the lipid solution, and extrude at least 11 times.
- The extruded sample from the acceptor syringe is withdrawn. The solution should look clearer than it was before extrusion.
- Cleaning
The lipid mixture of 1 mg/ml concentration contains 1, 2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1, 2-dioleoyl-sn-glycero-3-phosphoserine (DOPS) with 0%-10% DOPS volume. The solution is prepared with the organic solvent chloroform.
25 µl of sample solution are distributed as evenly as possible in the interior surface of a crystal meanwhile it is blown-dry using nitrogen. To totally evacuate the solvent of chloroform, the vial is left during one hour inside a drier.
500 µl of the working buffer (HEPES, NaCl and w/o Mg2+) is used to hydrate the dry lipid.
The vials are left for 2 hours of incubation; mix the sample using a vortexer.
The sample is immersed in liquid nitrogen followed by boiling water for 5 cycles totally.
Assemble the membrane inside the extruder as it follows:
The Teflon piece, o-ring and syringe should be rinsed with copious 2-propanol and deionized water after use. Otherwise the residues will contaminate the next sample.
Aptamer Lock
Optimization Experiments
- Equi-molar quanities of unlabeled aptamer strands and Cy3 labeled aptamer locking strands were mixed to produce samples with 1 nM, 10 nM and 100 nM concentrations of Cy3 labeled strands.
- All the samples were made in SLB buffer (<a href="http://openwetware.org/wiki/Biomod/2012/TU_Dresden/Nanosaurs/Lab_book#recipes">Lab Book -> Recipes</a>)
- The samples were then annealed in a PCR system and spectrophotometrically measured.
- Spectrophotometric measurements very done using 120 µl cuvettes in a spectrophotometer.
- The samples were excited at 510 nm and emission was measured at 564 nm.
Spectrophotometric Measurements
- All the spectrophotometric samples were prepared in SLB buffer (<a href="http://openwetware.org/wiki/Biomod/2012/TU_Dresden/Nanosaurs/Lab_book#recipes">Lab Book -> Recipes</a>)
- A 10 nM concentration of the respective labeled/unlabeld lock/oligo was used
- When PDGF was used, it was always used in 10 times excess concentration than the oligos
- When either of the blockers were used, they were used in a 10 times excess concentration than the oligos
- All samples were incubated at room temperature for 24 Hrs.
- Spectrophotometric measurements very done using 120 µl cuvettes in a spectrophotometer.
- All the samples were excited at 510 nm and emission was measured at 624 nm
Gel Shift Assays
- All the gel samples were prepared in SLB buffer (<a href="http://openwetware.org/wiki/Biomod/2012/TU_Dresden/Nanosaurs/Lab_book#recipes">Lab Book -> Recipes</a>)
- ~70 ng of DNA concentration was maintained for each sample.
- When PDGF was used, it was always used in 10 times excess concentration than the lock/oligos
- When either of the blockers were used, they were used in a 10 times excess concentration than the lock/oligos
- All samples were incubated at room temperature for 24 Hrs.
- Fermentas 6X Orange DNA Loading Dye was used with all the samples
- Fermentas O’RangeRuler 20 bp DNA Ladder was used
- Invitrogen, 4-20% TBE PAGE Gels were used and electrophoresis was performed for 40 minutes at 200 V
<a id="downloads">
Downloads
</a>
- <a href="http://biomod-dresden-2012.googlecode.com/svn/trunk/downloads/OligoList_for_DNA_Origami.xlsx">Oligo List for DNA origami</a>
- <a href="http://biomod-dresden-2012.googlecode.com/svn/trunk/downloads/shell_final.json">shell_final.json</a>
- <a href="http://biomod-dresden-2012.googlecode.com/svn/trunk/downloads/shell_final_guidestrands.json">shell_final_guidedstrands.json</a>
- <a href="http://biomod-dresden-2012.googlecode.com/svn/trunk/downloads/p7560_sequence_cadnano2.txt">p7560 scaffold sequence from cadnano2</a>
- <a href="http://biomod-dresden-2012.googlecode.com/svn/trunk/downloads/Lab_Book_DNA_Origami_group.docx">Lab Book of DNA origami group</a>
- <a href="http://biomod-dresden-2012.googlecode.com/svn/trunk/downloads/TEM%20guide.docx">TEM guide</a>
- <a href="http://biomod-dresden-2012.googlecode.com/svn/trunk/downloads/Staining%20Protocol.docx">Staining Protocol</a>
- <a href="http://biomod-dresden-2012.googlecode.com/svn/trunk/downloads/Agarose%20gel%20electrophoresis.docx">Agarose gel electrophoresis</a>
</body> </html>

{kind=link}
{kind=link}