Biomod/2011/TeamJapan/Sendai/Results/Electrophoresis
<html>
<style rel="stylesheet" type="text/css">
.clear {clear:both;}
#verticalmenu {
/*this .CSS is inspired by http://www.javascriptkit.com/ */ font-family: "Comic Sans MS" , "Brush Script MT",serif, sans-serif, monospace, cursive, fantasy; list-style:none;}
- verticalmenu a:hover {
color: #aa1d1d; /* color when the click is over the main menu text-transform: uppercase; font-size: 10px; */
}
a:visited { color:#00a5ea; text-decoration: none }
.glossymenu, .glossymenu li ul{ list-style-type: none; margin: 0; padding: 0; width: 250px; /*WIDTH OF MAIN MENU ITEMS*/ border: 1px solid black; list-style:none; }
.glossymenu li{ position: relative; }
.glossymenu li a{ background: white url(http://openwetware.org/images/a/a7/Glossyback2.jpg) repeat-x bottom left; font: bold 17px Verdana, Helvetica, sans-serif; color: white; display: block; width: auto; padding: 10px 0; padding-left: 10px; text-decoration: none; }
.glossymenu li ul{ /*SUB MENU STYLE*/ position: absolute; width: 200px; /*WIDTH OF SUB MENU ITEMS*/
left: 0; top: 0; display: none; }
.glossymenu li ul li{ float: left; }
.glossymenu li ul a{ width: 190px; /*WIDTH OF SUB MENU ITEMS - 10px padding-left for A elements */ }
.glossymenu .arrowdiv{ position: absolute; right: 2px; background: transparent url(http://openwetware.org/images/6/69/Arrow.gif) no-repeat center right; }
.glossymenu li a:visited, .glossymenu li a:active{ color: white; }
.glossymenu li a:hover{ background-image: url(http://openwetware.org/images/5/50/Glossyback.jpg); }
/* Holly Hack for IE \*/
- html .glossymenu li { float: left; height: 1%; }
- html .glossymenu li a { height: 1%; }
/* End */
</style>
<script type="text/javascript">
/***********************************************
- CSS Vertical List Menu- by JavaScript Kit (www.javascriptkit.com)
- Menu interface credits: http://www.dynamicdrive.com/style/csslibrary/item/glossy-vertical-menu/
- This notice must stay intact for usage
- Visit JavaScript Kit at http://www.javascriptkit.com/ for this script and 100s more
- /
var menuids=new Array("verticalmenu") //Enter id(s) of UL menus, separated by commas var submenuoffset=-2 //Offset of submenus from main menu. Default is -2 pixels.
function createcssmenu(){ for (var i=0; i<menuids.length; i++){
var ultags=document.getElementById(menuids[i]).getElementsByTagName("ul")
for (var t=0; t<ultags.length; t++){
var spanref=document.createElement("span")
spanref.className="arrowdiv" spanref.innerHTML=" " ultags[t].parentNode.getElementsByTagName("a")[0].appendChild(spanref)
ultags[t].parentNode.onmouseover=function(){
this.getElementsByTagName("ul")[0].style.left=this.parentNode.offsetWidth+submenuoffset+"px"
this.getElementsByTagName("ul")[0].style.display="block"
}
ultags[t].parentNode.onmouseout=function(){
this.getElementsByTagName("ul")[0].style.display="none"
}
}
}
} if (window.addEventListener) window.addEventListener("load", createcssmenu, false) else if (window.attachEvent) window.attachEvent("onload", createcssmenu) </script>
|
<img src="http://openwetware.org/images/1/13/Electroh.png" width="650">
|
</html> <html> <head> <style type="text/css">
- content {padding-left: 10px;width: 970px;}}
h3 {font-decoration: none;} h1.firstHeading {display: none; } </style> </head> </html>
<html> <style rel="stylesheet" type="text/css"> /*このスタイルシートの著作権はテンプレート工房TAKEにあります*/ /*ページのレイアウト用css*/
body{ background:#F5F5DC; /*壁色と壁紙設定*/ background-repeat:repeat;/*繰り返さない場合はno-repeatに変更*/ font:"メイリオ", "MS Pゴシック", Osaka, "ヒラギノ角ゴ Pro W3"; color: #333333; margin:0px; padding:0px; }
- contents{
width:900px;
margin:0 auto; background-color: #FFFFFF ;/*コンテンツ内の背景(サイズをぴったりにすること)*/ background-repeat:repeat-y; /*縦に繰り返し*/ border:solid 1px #666666;/*サイトに枠を付ける設定,色の変更可*/
position:relative;
font-size:80%;
}
/*ヘッダー部分の設定*/
- header{
background-image:url(http://openwetware.org/images/c/c4/TeamSendai-logo3.png) ;/*ヘーダー*/ background-repeat:repeat-x; /*縦に繰り返し*/ background-position:top right; height:140px; /*ヘーダーの高さ*/ }
- header p {
font-size: 26px;
color:#333333;
padding-top: 15px; padding-left: 20px; }
/*上部メニューボタンの設定*/
- navbar{
background-color:#FFFFFF;
width: 100%;
height:40px;
position:margine;
top:100px;
left:0px;
border-top:solid 1px #FFFFFF;
border-bottom:solid 1px #FFFFFF;
}
- navbar ul{
margin:0;
padding:0; list-style-type:none; font-family:Arial, Helvetica, sans-serif; font-size: 12px; line-height:40px; letter-spacing:2px; }
- navbar li{
background-color:#000099; /*上部メニューのボタンの背景*/
float:left; width:146px; /*メニューボタンの幅*/ text-align:center; padding:0; border-right:solid 1px #ffffff; }
- navbar ul a:hover{
background-color:#0033cc; /*メニューボタンにカーソルが来た時に背景*/
width:146px; /*メニューボタンの幅*/ }
- navbar a{
color:#ffffFF;/*メニューボタンの文字の色*/
display:block; }
- navbar a:hover{
color:#999999; /*メニューの文字がカーソルが来た時、この色に変わる*/ }
/*サイドメニューの設定*/
- side{
background-color:#ddffff;
width:220px;/*サイドの幅(変更するときはコンテンツ背景も変更すること)*/
position:margin; top:600px;/*上からの位置*/ left:12px; }
- side h3 {
font-size: 90%; border: double 3px #FFFFFF; color:#ffffff; text-align: center; background-color:#999999;
width:190px;
line-height: 30px; margin-top: 10px; margin-left: 5px; margin-bottom: 5px; }
- side h3 a {
color:#ffffff;
font-weight:normal; }
- side ul{
font-size:100%;
line-height:220%; /*サイドの文字と文字の行間設定*/ background-color: #ddffff; margin:0px; padding-left:10px; }
- side ul a:hover {
width:180px;
background-color: #99ffff; /*サイドのカーソルオーバー時の背景色*/ color: #999999; /*サイドのカーソルオーバー時の文字色*/ }
- side ul{
list-style-type:none;
padding-left:2px; }
- side li{
padding-left:15px; /*文字の左端からの位置*/ }
- side li a{
color:#333333;/*サイドの文字色*/
width:180px;
display:block;
}
- side .ad_list li{
background-image:none;
padding-left:0; }
/*右側メイン部分の設定*/
- main{
width:630px;
padding-top:15px;
margin-left:240px;
}
/*下部のフッター部分の設定*/
address{
font-size:80%;
font-style:normal;
text-align:center;
padding-top:5px;
}
address{
background-color:#000066;
color:#ffffff;
width:882px;
padding-bottom:10px; border:none; } address a{
color:#ff9999;
}
/*文字の設定*/ h1{ font-size:60%; letter-spacing: 2px; padding-left:10px; margin: 0px; }
h1 a{
color:#FFFFFF;
font-weight:normal; }
h2{
font-size:140%;
border-left: 10px solid #000066;
border-bottom:solid 1px #000099;/*文字の下に線を入れる設定*/
width:900px;
padding-left: 5px; color:#333333; margin-top: 15px; margin-bottom: 5px; }
h3{
font-size:120%;
border: solid 1px #111111;
color:#ffffff;
background-color:#4682B4 ; line-height: 30px; padding-left:10px; margin-top: 10px; margin-bottom: 1px; }
p{
font-size:90%;/*全体の文字サイズ*/
line-height:150%;/*全体で使う、文字と文字の行間*/
margin-left:5px;
}
p img{
float:left;
margin-top:5px; /*写真の左にスペースを空ける*/
margin-left:5px; /*写真の左にスペースを空ける*/
margin-right:10px; /:写真と文字の間隔*/ }
/*リンク文字の設定*/
a{
color:#000099;
text-decoration:none;
} a:hover { color: #FF0000;/*リンクの文字の上にマウスが来た時この色に変わる*/ text-decoration: none; }
- purple{
font-size:120%;
border: solid 1px #111111;
color:#ffffff;
background-color:#9370DB; line-height: 40px; padding-left:10px; margin-top: 10px; margin-bottom: 1px; }
h5{
font-size:120%;
border: solid 1px #111111;
color:#ffffff;
background-color:#FFA500; line-height: 30px; padding-left:10px; margin-top: 10px; margin-bottom: 1px; }
h6{
font-size:120%;
border: solid 1px #111111;
color:#ffffff;
background-color:#006400; line-height: 30px; padding-left:10px; margin-top: 10px; margin-bottom: 1px; }
- red{
font-size:120%;
border: solid 1px #111111;
color:#ffffff;
background-color:#DC143C; line-height: 40px; padding-left:10px; margin-top: 10px; margin-bottom: 1px; }
- blue{
font-size:120%;
border: solid 1px #111111;
color:#ffffff;
background-color:#191970; line-height: 40px; padding-left:10px; margin-top: 10px; margin-bottom: 1px; }
</style> </html>
About the methods
In this section we are presenting the procedures we took for the following cases:
- Verifying 2D and 3D structures. Basically we verified whether the folding was properly done or not.
- Verifying whether the DNAzyme legs cut the substrates. Important step for the robot motion.
- After the folding, there were some leftover staples. We needed to get rid of them before the next step.
- Because we cut M13 it is needed to check whether we got the desire 1,108 nucleotides.
Take into consideration that DNA strands were stained by SYBR Gold (sigma) except when observing them with fluorescence.
<html>
|
|
</html> <html>
|
|
</html>
Method for verifying the structure
Verifying 2D and 3D structure (using whole M13 for the robot body)
Experiment conditions:
- Gel: 1.4% agarose gel
- Buffer: 1×TAE Mg2+
We tested the influence of time and ratio to make our 2D and 3D nanostructures, by changing annealing time and concentration ratio between M13 and staples. This staples are the needed for forming 2D and for folding the 2D structure into the 3D structure. From now on, the last kind of staple will be refer as 'staples for forming the 3D structure'. As a result of the experiment, we got that the 2h annealing was better than 3h annealing, and a high concentration of staples (1:100) was better.
 |
 |
Verifying the 2D and 3D structure (using cut M13)

We carried out electrophoresis to 2D and 3D structure to figure out how the band gap differences among these two are. Figure 3 shows the band gap of 2D and 3D structure.
To examine whether we succeed to construct the designed 2D DNA origami, electrophoresis were performed. Folded 2D and 3D structure were observed as migrations in agarose gels. Figure 3 shows different migrations of 2D and 3D structure by the electrophoresis.
Experiment conditions:
Cut M13: 58.8 nM for each 3.4μL
Staples: 400 nM for each 5μL
5×TAE-Mg2+: 10μL
Distilled water: 31.6μL
Total: 50μL
Annealing: from 90ºC to 25 ºC (decreasing 1 ºC each 3 minutes)
As a result of the electrophoresis: Band C indicates the cut M13. Band B indicates the 2D structure, because this band shows an slower migration than cut M13. Two bands were observed in lane #4, and the lower band showed similar migration as the band in lane #3. There are two possibilities to consider in case of the upper band. First, one of the two bands indicates the band of the triangular prism structure. Second, this is the band which was made by connection of 2D structures between each other. However, if the upper band indicated connection of two pieces of 2D structures, two or more bands should have been observed. Thus, we arrived to the conclusion that the upper band indicates the designed 3D structure. Our AFM observation supports this conclusion. Please see this page for the AFM observation.
Method for verifying that the DNAzyme legs cut the substrates
DNAzyme Legs and a substrate with internal ribonucleotide and fluorescence protein at 5’ end
Experiment conditions:
- Gel: 24% Poly-Acrylamide Gel including 6M Urea
- Buffer: 1×TAE
- Sample: mixtures of DNAzyme Legs and the substrates
In figure 3, samples loaded in each well are:
1 and 5 contain 1xTA
2 and 6 contain 1xTA Mg2+ (12.5mM)
3 and 7 contain 1xTA Zn2+ (1mM)
4 and 8 contain 1xTA Mg2+ (12.5mM) and Zn2+ (1mM)
Where #1-#4 are immediately loaded after mixing, and #5-#8 are loaded after incubation during 15 min at room temperature. Electrophoresis showed bands which migrated faster than original substrates. The faste migrated bands indicate cleaved substrate, and that were observed in samples with cations (lane #2, #4, #6, #8). Amount of cleaved substrates increased by 15 min incubation. Figure 4 shows that differentiation between lane #2 and #6 (red line), and between #4 and #8 (green line) substrates, showing Zn2+ enhanced cleaving efficiency.
 |
 |
Cleaving of substrates without fluorescence by DNAzyme legs

Experiment conditions:
- Gel: 24% Poly-Acrylamide Gel with 6 M urea
- Buffer: 1×TAE
- Sample: Legs and substrates are loaded on line 1, 2, 3 and 4. Substrates and DNAzyme legs were loaded on 5 and 6, respectively.
Mg2+ (12.5 mM) Zn2+ (1mM), Zn2+ (1mM), and Mg2+ (12.5 mM) were added to the sample of lane in 1, 2 and 3, respectively.
After 15 min incubation, samples were loaded on agarose gels. Figure 5 shows that substrates of 1, 2 and 3 are properly cut, indicating that the double helix was formed.
Method for removing remained staples after DNA origami folding
Gel filtration by a micro spin column S-400HR

Experiment conditions:
- Gel: 0.7% agarose gel
- Buffer: 1×TAE Mg2+
To remove remained staples after the DNA origami fold, a gel filtration was performed using S-H400R micro spin column (GE health care). Through gel filtration, remained staples were totally removed (see figure 5). However, folded structures also diminished. Since folded structure was not observed by AFM, we concluded that the folded structures disrupted through SH400R gel filtration.
polyethylene glycol (PEG) precipitation

We carried out a PEG precipitation to get rid of the leftover staples from the sample.
Experiment conditions:
- 10mM MgCl2, PEG6000 10%, to a sample.
- At 16,000×g in a conventional tabletop microcentrifuge for 10 minute at RT.
- Removed Supernatant liquid and a sample is dissolved by 1×TAE-Mg2+ of 80% of a total amount.
It seemed that more staples were removed by two consecutive PEG precipitation.
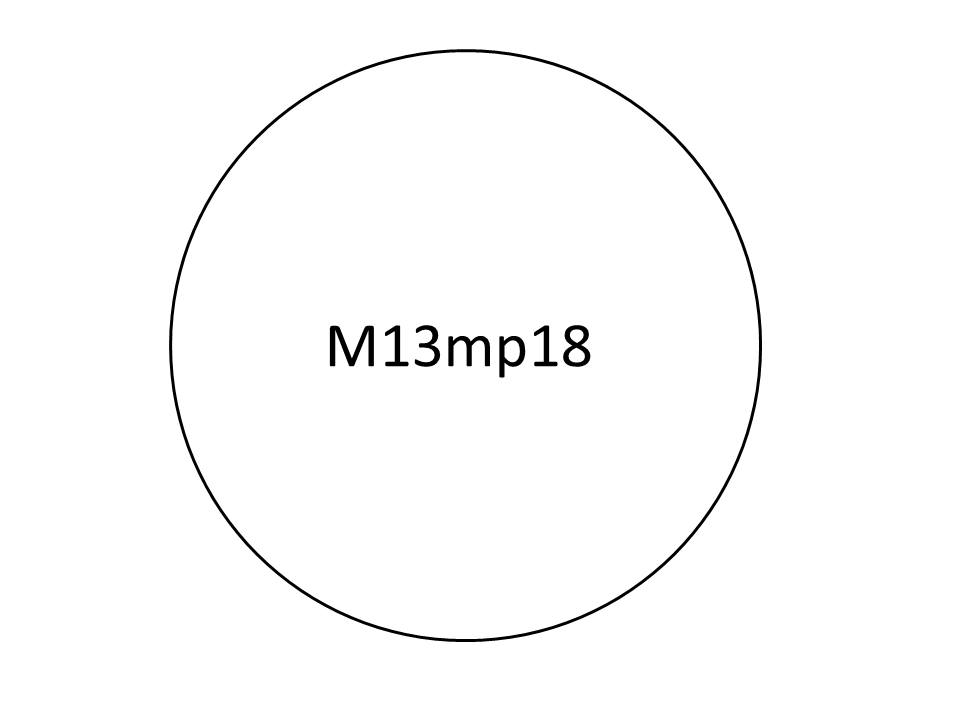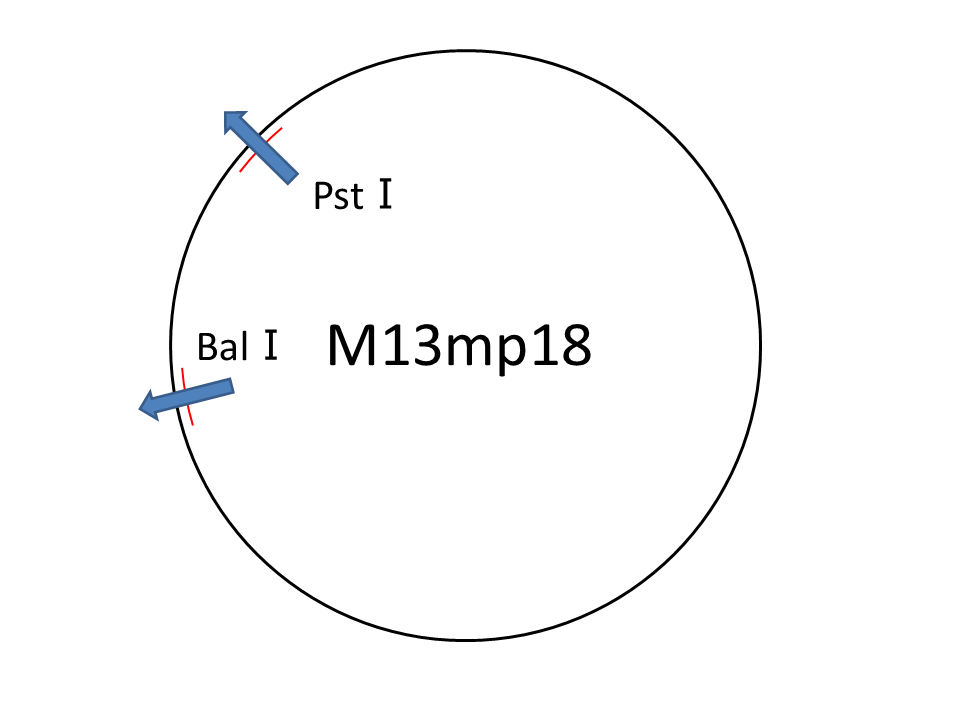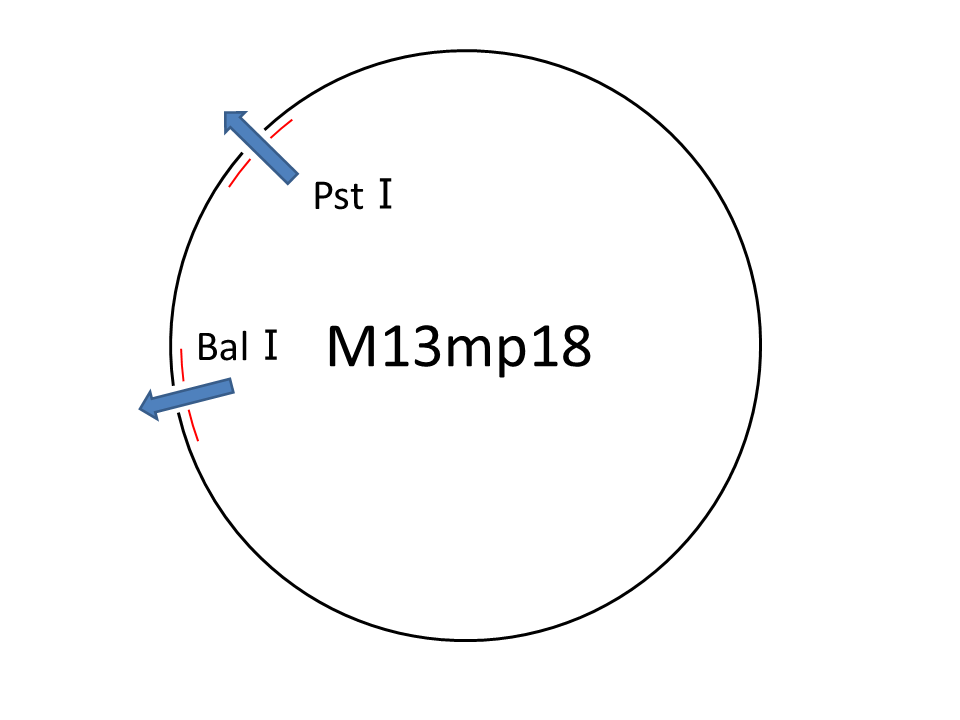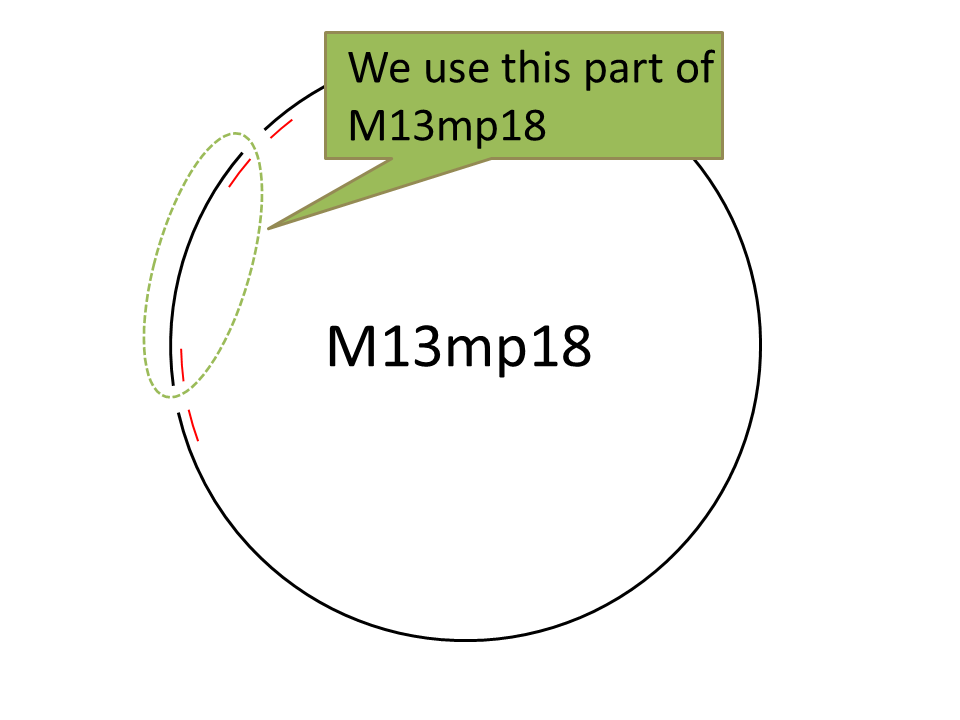
Method for cutting M13

Experiment conditions:
- Gel: 1.0% agarose gel
- Buffer: 1×TAE
For producing the robot body, we firstly used whole M13mp18 DNA single strand (M13) as scaffold.
However, 1,108 of 7,249 nucleotides were used in our design (just 16% of the total M13 strand). Thus, the region no necessary to be used was cut with restriction enzyme (Bal I and Pst I). In figure 6, different degrees of migration of the whole M13 and cut M13, after treatment with restriction enzymes, were compared by electrophoresis. Label 1: shows the whole M13, and label 2: the M13 after the cutting treatment. Band A: the band corresponding with the whole M13. Band B: the band corresponding to the longer cut M13 treatment with restriction enzymes. Band C: the band corresponding to the shorter M13 which we use in our DNA origami. Band D: the band of the short DNA (20base) which was used to form partial double helix to cut M13 by restriction enzymes. The reason why the band C is slower than D is because the samples were purified by the QIAGEN PCR purification kit, by which long DNA strands (more than 5000base) are not effectively purified, before loading gels. Detailed procedures are described in Here.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}