Biomod/2011/Harvard/HarvarDNAnos:LabNotebook Sherrie
<html> <head>
<style>
- column-one { display:none; width:0px;}
.container{background-color: #f5f5f5; margin-top:50px} .OWWNBcpCurrentDateFilled {display: none;}
- content {width: 0px; margin: 0 auto auto 0; padding: 1em 1em 1em 1em; align: center;}
- column-content {width: 0px; float: left; margin: 0 0 0 0;padding: 0;}
.firstHeading {display:none; width:0px;}
- globalWrapper{width:1280px; margin:auto}
body {background: #F0F0F0 !important;}
- column-one {display:none; width:0px;background-color: #f0f0f0;}
- content{border:none;margin: 0 0 0 0; padding: 1em 1em 1em 1em; position: center; width: 800px;background-color: #f0f0f0; }
.container{ width: 800px; margin: auto; background-color: #f0f0f0; text-align:justify; font-family: helvetica, arial, sans-serif; color:#f0f0f0; margin-top:25px; }
- bodyContent{ width: 1267px; align: center; background-color: #f0f0f0;}
- column-content{width: 1280px;background-color: #f0f0f0;}
.firstHeading { display:none;width:0px;background-color: #f0f0f0;}
- header{position: center; width: 800px;background-color: #f0f0f0;}
- footer{position: center; width:1280px;}
</style>
</head> </html>
<html>
</html>
Sherrie's Lab Notebook
Week 1
Monday (2011-06-06)
Dear Lab Notebook,
- First day of BioMod at the Wyss Institute! This place is well furnished; I'm especially a fan of the huge sliding glass doors. Anyway, this morning we had a brainstorming session with Adam concerning box designs. We came up with three different designs. The first is a cubical box constructed with helices all going in one direction, two halves of which are apart but connected by scaffold when open and together when closed. The closing mechanism could be a tweezer design, and the lock could be opened via strand displacement. The second design is a box with helices again going in the same direction, on one face of which there is a hole closed by a mesh of staple strands binding to each other. We could design the staple strands so that the ends closing the hole all have the same sequence and can be displaced by the same key strand or cut by the same restriction enzyme. The third idea is a spherical box. We're not sure yet how to construct this sphere or how to open it, but we like the idea because it uses DNA efficiently. The box-with-helices-all-going-the-same-direction design is good because it has no seams and therefore few points of weakness, but it uses DNA rather inefficiently. Adam also suggested using 5nm gold particles as our cargo.
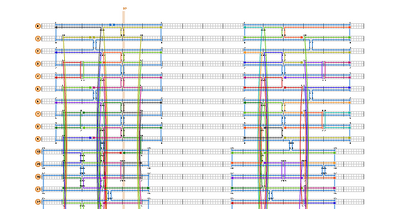
- I started working on the first box design; below is a screenshot of the file in caDNAnoSQ.
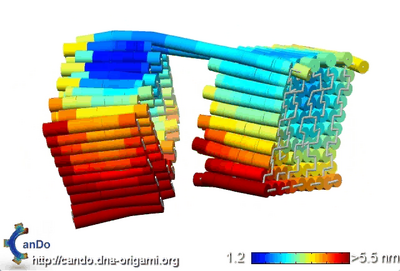
- When sent into CanDo, I got back a video showing thermal fluctuations of >5.5nm. That's expected, since the sides of the box not connected by scaffold are free to swing sideways. The fluctuations between helices look pretty small, so the box shouldn't leak 5nm gold particles between helices. I'm not sure if the scaffold between the two halves is long enough; when the box is in its closed state, will the scaffold bend and allow the box to close seamlessly, or will it resist bending and cause the box to have a gap? Below is a screenshot of the CanDo video.
- Here's the caDNAnoSQ file I made at the end of the day.
- Media:Box2.json


- Sherrie's Aarhus box: CanDo gives bad input file error
Tuesday (2011-06-07)
Dear Lab Notebook,
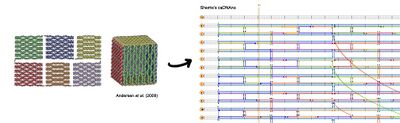
- This morning, I worked on reconstructing the Andersen et al. box from a 2009 paper published in Nature. It's a box made of six roughly square sides folded together, as shown below. I tried to figure out why the Andersen box is not suitable as a robust container; there are a lot of seams, but can't we just staple them shut?
- I soon realized, though, that there are some difficulties with stapling one side to another. Crossovers don't often match, helices are running in different directions, etc. I had a hard time figuring out where to force crossovers, especially for edges where the helices run perpendicular to each other. I'm not sure at this point if this box is worth pursuing.
- In the afternoon, we had another brainstorming session, this time also with Tom and Wei. With Tom we talked a lot about opening and closing mechanisms for boxes. Since the distance between bases is 0.43nm in ssDNA and 0.34nm in dsDNA, he suggested using the hybridization of staple strands hanging off the lid to those hanging off the box to apply force to the lid when it's closed. He also suggested a spring mechanism to open the box. Wei informed us that 5nm gold particles aren't just 5nm in solution; they're effectively larger due to a hydrodynamic shell. So we should design our box to have an opening much larger than 5nm in diameter; Wei said at least 20nm.
- Food for thought.

Wednesday (2011-06-08)
Dear Lab Notebook,
- Worked all day on the box design suggested by Tom yesterday. As of now it's got one-helix thick walls, a one-helix thick bottom, and a two-helix thick lid. Here's a caDNAnoSQ screenshot of the box.

- I ran into trouble when the box I initially designed had too many bases for the M13 sequence. I had to redesign the box a couple of times, making sides smaller and the box a bit shorter. It doesn't use DNA efficiently, which makes this design not so elegant. I'm not sure how to incorporate a spring opening element as Tom suggested; it seems I would need to make the box bigger to accommodate that, yet I'm constrained by the M13 sequence.
- Later in the day we decided to embark on quest to reconstruct sphere from Han et al.'s paper in Science. It looks challenging and really interesting; we'll see how this goes!

Thursday (2011-06-09)
Dear Lab Notebook,
- I finished a draft of the lidded box design suggested by Tom. Adam had another idea for the lid: to use stacking interactions to help hold the lid on the box. I wonder if the lid will still come off easily though? Here's the caDNAnoSQ file.
- Media:Box3.json
- Below is the CanDo file that came back with thermal fluctuations of the box. I'm not sure what happened with the lid; where I've attached it to the scaffold is contorted weirdly. CanDo says there are very small thermal fluctuations, though, which is great.

- Then I started working on reconstructing the sphere from Han et al.'s supplemental materials. It's not as bad as I initially thought, especially since they did all the calculations for us of how many bases should be in each ring layer of the sphere, where crossovers can happen, etc. It's a matter of accurately reconstructing their design. It took me almost all day, but I finally have a scaffold of the sphere done. Adam and I took a bit of time to figure out how to construct the southern hemisphere of the sphere and connect it to the northern hemisphere, since the paper only provides instructions for designing one hemisphere. We decided a mirror image should work.

Friday (2011-06-10)
Dear Lab Notebook,
- It's Friday! Gotta get down.
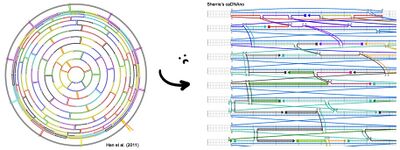
- Continued construction of the sphere in caDNAno today, manually placing all the staple strands in the exact positions Han et al. diagrams them in Figure S10 (see below). Unfortunately, caDNAno did not make this easy. Figure S10 provides the exact nucleotide positions where staple crossovers occur; to find these positions in caDNAno, I have to use the Slide Bar in a very slow and arduous process. After five hours, I finally have the northern hemisphere all stapled. Only one more hemisphere to go...
- In other, more uplifting news, I downloaded Maya and have started watching the short video tutorials that come with it. We plan to make a 3D model of the DNA origami sphere and write a Python script to tell us the caDNAno coordinates of any base in the 3D model. The goal is to be able to model more intuitively in 3D and then have the script translate the 3D model into caDNAno. Much more friendly than figuring the crossovers out on paper! I'm looking forward to playing around with Maya to make the model.

Week 2
Monday (2011-06-13)
Dear Lab Notebook,
- Finished the staples for the southern hemisphere, then connected the two hemispheres!! The design looks quite nice zoomed out; Evan says it's like a peacock feather. Take a look!

- Before connecting the two hemispheres, I sent the file into CanDo just to see what would happen. It doesn't look like two halves of a sphere, but if you squint and tilt your head to the side...
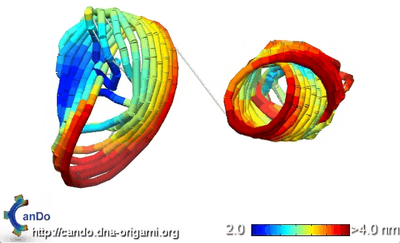
- Still doesn't look like two halves of a sphere. Oh, MIT.
- Now I'm working on matching all my staple strands with Han et al.'s staple strands. Adam wrote a Python script to do this, and the first run was able to match 120 out of 160 staples. After I connected the two hemispheres, that number was up to 136. Adam modified the script to output which strands don't match, and from that I was able to fix some. It turns out that Han et al. didn't make all the staple strands cross over as pictured in Figure S10, which is misleading. There are also some places where bases simply don't match, e.g. there is a G in the middle of a staple strand where caDNAno says there's an A. I'm up to 140 matches right now, with the rest being these crossover or random base mismatches.
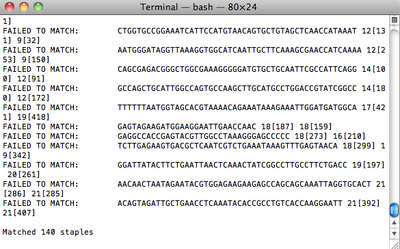



Tuesday (2011-06-14)
Dear Lab Notebook,
- Continued fixing mismatched staples this morning. Got the number of staple matches up to 142, with 16 staples not matching due to seemingly random base differences (A instead of T) nowhere near crossovers. Not sure why these exist. Also weird is that 142 and 16 add to 158, which is two staples short of the 160 staples Han et al. ultimately used in their sphere. Where'd they go??
- Here's the file with all the mismatches thus far.
- Media:Sphere_mismatches.rtf
- In the afternoon, I fumbled around some more with Maya and followed a tutorial to make a screwdriver! Things went pretty smoothly until I had to render using mental ray... I couldn't get the dgs_material and dielectric material of the screwdriver to render properly; they just turned up black. I spent a couple hours tweaking settings, but to no avail. That's alright, though, I learned about selecting vertices, moving stuff, duplicating stuff, scaling stuff, etc. etc. Here's what the screwdriver looks like.


Wednesday (2011-06-15)
Dear Lab Notebook,
- This morning I lost my wallet and ran around calling banks and canceling cards. Fun fun fun.
- More relevantly, I modified the staple-checking script to output all the staples that match as well as those that don't match. From this output, I color-coded the caDNAno file, making all matches black and all mismatches red. As expected, the matches and mismatches together accounted for all my staple strands. But that's still only 158; where are the other two?
- Turns out there ARE only 158 staples. Not 160. Thanks, Han et al. All the staple strands are numbered North or South from 1 to 80, so you'd think there are 160 of them, right? Nope, not when the 14th strands don't exist at all. So the sphere I made has the right number of staples after all.
- Okay, big picture now. Time to think about what to do with the sphere. A possible opening mechanism is getting rid of the staples holding together the two hemispheres and replacing them with a few staple strand locks that can be displaced. How do we close the hole at the top and bottom, though? A few staples crossing over the top? A cap of some sort? How big even is the hole?
- We also imaged some long DNA origami rectangles with Ralf. We mixed staples, M13 scaffold, and folding buffer and put it all in the thermal cycler, then used the AFM to image it. Ralf and Adam also explained some AFM principles to us.
- Continued working with Maya, this time trying to construct a DNA helix. It'd be cool if we could get helix to wind around a sphere in our Maya model.
- Here's the sphere as of now, with staples matching Han et al.'s colored black and staples not matching theirs colored red.
- Media:Sphere.json
- Since there's no picture in today's entry, here's a view of what our workspace looks like:

Sherrie's sphere: Media:Sphere.json
- Note: Black staples match those of Han et al. while red ones do not (for thus far unknown reasons)
Scaffold Permutation Analysis Python script used to determine if our produced staples match those provided by the literature: Media:Sphere debug.zip
Thursday (2011-06-16)
Dear Lab Notebook,
- This morning I worked on designing locks along the equator of the sphere. I used Nick's excel spreadsheet to find all the bases pointing outward, which is where I can put the locks.
- In the afternoon, the team met with Peng Yin, William Shih, and other lab members to discuss our progress and how to proceed. We talked a lot about ways to solubilize the cargo inside the box. Shih suggested that we use disulfide bonds, which are stable at high temperatures and can be cleaved with the addition of DTT to solution, to connect the cargo to the inside of the box. We can also use strand displacement; it's possible that single strands can diffuse inside the sphere (though slowly) through the top and bottom holes. Azobenzene is another option, as is restriction enzymes. Not sure if restriction enzymes will diffuse through ~4nm holes though.
- We also talked about another interesting design for a box that we can explore later: a torus! The nice thing about a torus is that it's a totally closed space. This idea applied to the sphere we have is just to run DNA rings through the center of our sphere to close it off. Or, maybe we can make a cylinder of DNA rings, then bend them to form a torus, then cut the torus at one ring to release cargo.
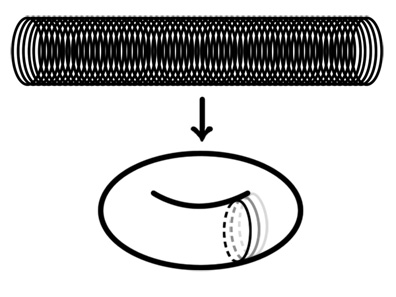
- List of things we want to construct:
- Normal sphere
- Sphere with disulfide on equator and cargo handle staples
- Sphere with strand displacement cargo handle staples, disulfide on equator
- Sphere with strand displacement equator staples, disulfide handle staples
- Sphere with restriction enzyme staples
- Sphere with azobenzene staples
- Cubical box

Friday (2011-06-17)
Dearest Lab Notebook,
- Created a spreadsheet with "equator" staple sequences for a disulfide sphere and a strand displacement sphere.
- Media:Sphere_staples.xlsx
- To do this, I used Nick's spreadsheet to find where staple strands along the sphere's equator face outward on the northern and southern hemispheres, then where these outward strands line up. I also designed tweezers with staple strand extensions, a lock strand, and a key strand complementary to the lock strand. The current sequence I have is taken from the Liang paper, so I assume it will work better than something I randomly generate (but maybe not). I did, however, have to come up with my own addition to the lock and key strands, as the Liang paper doesn't use strand displacement to open the tweezers. Here are the NUPACK results of the lock and key, respectively.
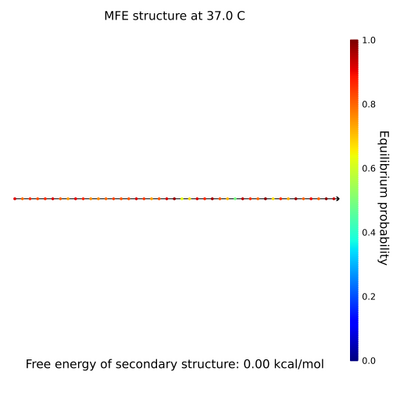
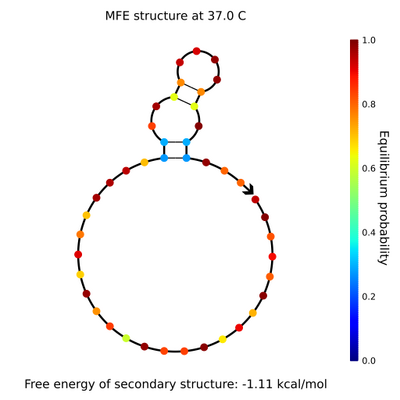
- The staple extensions are universal; that is, the same staple extensions will be added to the ends of all staples on the equator so they can be locked with the same lock and opened with the same key. It'd be interesting to explore different tweezer sequences and see which ones hold the sphere closed best, which ones are easiest to open, etc. It'd also be neat to vary the number of locks holding the sphere closed; is 2 enough? How long will it take 9 to open?


Week 3
Monday (2011-06-20)
Ahoy there Lab Notebook,
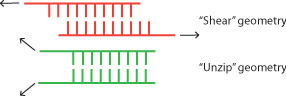
- This morning we talked to Tom about the forces that hybridization of DNA strands can provide on an origami box. According to this paper, the force exerted by a DNA lock in the shear geometry (see below) is ~50 pN. I'm not sure how big this is for a DNA origami box, but according to Tom we can calculate how much 50 pN will cause a DNA lid to bend using the same equations engineers use for buildings, which is pretty cool.
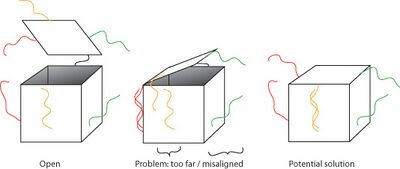
- Tom also brought up a potential problem for assembling the lid: if one lock hybridizes first, it may separate the other sides of the lid from the box such that the other locks can't lock. Then all our boxes will be open, because it's almost guaranteed that one lock hybridizes before the others. One potential solution is to design the lock opposite the lid hinge such that it hybridizes first; another solution is to add the opposite lock strand first, then the side lock strands.
- In the afternoon I made a schematic of the strand displacement/tweezer method for opening the sphere.
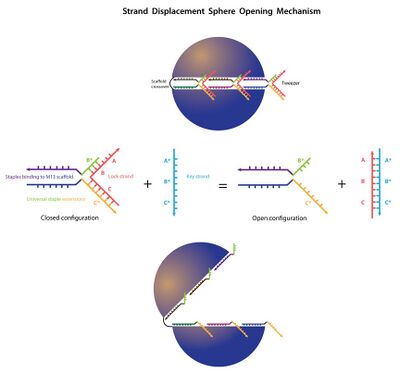



Tuesday (2011-06-21)
Dear Lab Notebook,
- We started experiments today! In the morning we diluted the DNA we're using for disulfide cross-linking and gold nanoparticle conjugation.
- In the afternoon, Steve showed us how to use the dynamic light scattering (DLS) machine to figure out the diameter of gold nanoparticles. The stock of 5nm gold particles that Adam has showed some aggregation of nanoparticles (it's been in a fridge for a while) with hydrodynamic diameters of 50nm and 5um. Here's the data file the machine output:
- Media:Gold_DLS.txt
- Later, Evan and I continued diluting the DNA (oh yay stoichiometry) and mixed an M13 ultramer with an increasing number of oligonucleotides to create strands of DNA off which hang oligonucleotides with tails. The goal is to eventually attach gold nanoparticles to this strand and get a "necklace" of sorts.
Wednesday (2011-06-22)
Dear Lab Notebook,
- This afternoon we made our own 5nm, 15nm, and larger gold nanoparticles with Steve. You just heat gold citrate and add stabilizer and reducing agent (citrate and tannic acid/citrate depending on the size of the particles desired); pretty simple! They came out pretty well, we think. They also came out pretty, period.

- We then used dynamic light scattering to characterize the nanoparticles, and you can see below the hydrodynamic diameters of the nanoparticles.
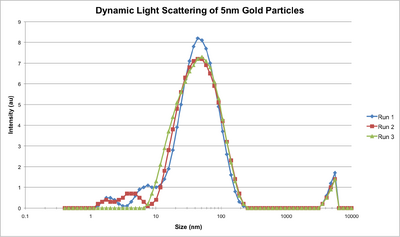
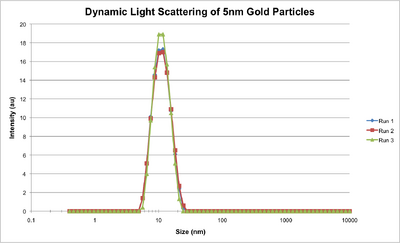
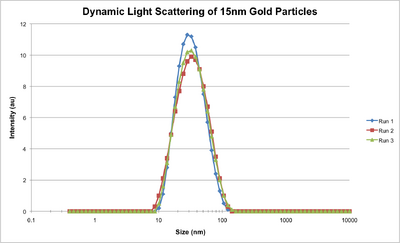
- We also ran a gel with our ultramer and increasing numbers of handles, but the gel broke the first time so we had to do it again. The second try came out well, though; you can see below the shift in bands as the ultramer has more handles connected to it. We didn't run it for enough time so the bands haven't quite separated, so we'll do it again.
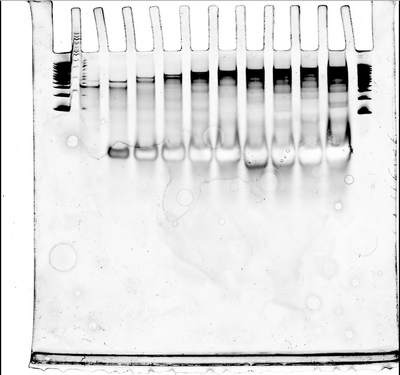
- Also. Nick is a mad scientist.



.png)



Thursday (2011-06-23)
Dear Lab Notebook,
- Spent the morning concentrating gold nanoparticles and working intermittently on a powerpoint presentation to be given at the Yin lab meeting. Finished the powerpoint in the afternoon and presented first at lab meeting. Some useful feedback:
- Thiolated nanoparticle strand needs to be distinct from the disulfide
- Strand (to allow conjugation to gold without breaking the disulfide - gold itself can competitively reduce disulfides)
- Box foldings with AuNP cargo should be 2-step not 1-step
- Linking of 5' S-S strand directly to gold particle works with high efficiency. We have one of these for the 15-mer, but it is short, so gel separation won't be good. Still, we can try it
- Look into conditions necessary for restriction enzyme activity
- Caged DNA instead of azobenzene: impractical this summer since we have the azobenzene strand already
- Use type-II restriction enzyme which is an "offset cutter" to cut close to origami while keeping cut site a reasonable distance away from the origami
Friday (2011-06-24)
Dear Lab Notebook,
- This morning we reran an ultramer-handle band shift gel and, now that we know the handles are binding to the ultramer, also ran a gel containing lanes with the ultramer and all the handles. The latter gel is for purification of ultramers containing all the handles.
- Things to test for strand displacement opening mechanism:
- Number of locks needed to close sphere (find minimum)
- Amount of time it takes to open sphere with N locks
- Tweezer sequences (find type [A/T? C/G?] with highest yield for opening/closing)
- Tweezer lengths (what range to test?)
Week 4
Monday (2011-06-27)
Dear Lab Notebook,
- Today we tried a gel extraction of the gold nanoparticles conjugated to DNA based on instructions from Steve. It didn't seem to work, though, as the gold nanoparticles migrated extremely slowly and not in distinct bands. I wonder what went wrong... We're gonna wait for Wednesday (when Steve gets back) to try again.

- I also finished up revising staples for the strand displacement opening mechanism, and now have a new document of sphere staples.
- Media:Sphere_staples_updated.xlsx
- In the afternoon, Nick and I figured out where we wanted to place our handles for loading cargo. Then I spent an annoyingly large amount of time sorting all the sphere staples into groups: those that are "Han" staples that are neither lock nor handle, those that are "my" staples, those that are "Han" lock staples, and those that are "Han" handle staples. Tomorrow I can start pooling the strands and hopefully folding the sphere!
- Media:Sphere_wells.xlsx

Tuesday (2011-06-28)
Dear Lab Notebook,
- This morning I pooled the sphere staple strands according to the document I created yesterday. In total there are now 28 pools: one with 109 staples that match Han's and aren't part of the lock or handle mechanisms, one with 16 staples that don't match Han's, 25 with locks, and four with handles.
- Then, in the afternoon, I made four reaction solutions: one using buffer with 12.5mM MgCl2, one using buffer with 12.5 MgCl2 and gold nanoparticles, one using buffer with 16mM MgCl2, and one using buffer with 16mM MgCl2 and gold nanoparticles. With the gold nanoparticles, we're curious just to see if the gold will be folded inside the sphere by chance, and what that yield is. The mixtures are currently in the thermocycler, following the cycle given by the experimental methods from the Han paper. In 12 hours we'll see if we have spheres!
Miércoles (2011-06-29)
Querido laboratorio portátil,
- Esta mañana, preparé un gel con las esferas. También les ayudé a Evan y Nick preparar geles para sus experimentos. Terminé las secuencias para las grapas con manejas; puedes ver la hoja de cálculo debajo.
- Media:Sphere_staples2.xlsx
- Usamos el AFM para obtener un imagen de las esferas. No es ideal para 3D origami, sin embargo podemos ver algunas formas circulares que esperamos que sean las esferas. Aquí está un imagen de "las esferas":

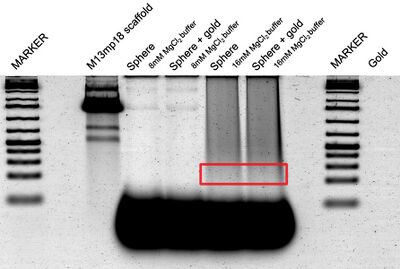
- Esta tarde, purifiqué las esferas con el gel; corté la sección roja en el gel (debajo), la trituré, y la filtré. Puedes observar en los lanes con 8mM MgCl2 buffer las rayas del M13 scaffold; no hay origami. En la ponencia de Han et al., se usó 16mM MgCl2 buffer. En los lanes con este buffer, puedes observar origami. No sé por qué hay muchas rayas en el lane de M13 scaffold... debe existir sólo una.
- Usé el AFM para obtener un imagen de estas esferas purificadas, y aquí está. No estamos seguro de que sean esferas o no.




First Folding of the Sphere (unsuccessful due to too-low buffer concentration)
Gel:
AFM of unpurified spheres:
AFM of purified spheres:
Thursday (2011-06-30)
Dear Lab Notebook,
- Did TEM imaging this morning with Wei. We're not sure if we saw spheres or just water droplets, though... The staining wasn't great so it was hard to tell. We did see gold nanoparticles, though; they're black under TEM. See for yourself.
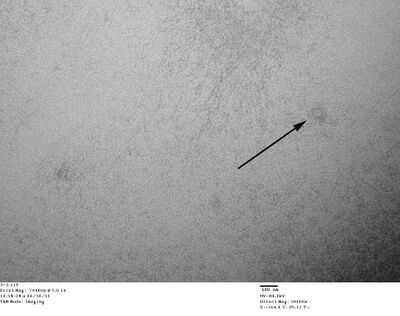
- Above there's a circular shape that is around the right size to be a spherical origami. Or just a drop of water.

- Here you can see the gold nanoparticles, around 5nm as they should be. The lighter circles look promising, but are too small to be our origami; they're ~20nm in diameter while our origami should be ~40nm.
- If we indeed did not see any spheres in the purified samples, possible explanations include:
- Purification did not work
- Staining was sub-optimal
- Spheres did not fold (we had 16 imperfectly matched staples, though this shouldn't prevent the whole structure from forming)
- The band we took from the gel did not contain spheres
- Things to try:
- Image unpurified spheres
- Stain unpurified and purified spheres again with Wei
- (If we see spheres in the unpurified sample but not the purified) Move our way up the gel, purifying different sections of the smear
- In the afternoon, we had a meeting with William Shih and Peng Yin, who reminded us that gold nanoparticles cleave disulfide bonds, meaning we probably can't use disulfide bonds as an opening/closing and solubilization mechanism. Instead they suggested a photo-cleavable linker, which we had previously considered but not pursued. I updated the inventory of sphere staples and designed new photo-cleavable equator staples and handles.


TEM of purified spheres. Do we see spheres or just water droplets?
Friday (2011-07-01)
Dearest Lab Notebook,
- Per Wei's suggestion, this morning I used the nanodrop to measure the concentration of origami in the purified and unpurified sphere samples. According to Wei, the A260 column should read around 0.15 or 0.2. This was indeed the case.
Sample A260 Reading Unpurified sphere 16.851 Purified sphere gel position 1 0.145 Purified sphere + AuNP gel position 1 0.135 Purified sphere + AuNP gel position 2 0.199 Purified sphere + AuNP gel position 3 0.151 Purified sphere + AuNP gel position 4 0.151 Purified sphere + AuNP gel position 5 0.170 Purified sphere + AuNP gel position 6 0.131 Purified sphere + AuNP gel position 7 0.160
- In the afternoon, we ran a gel of the boxwithlid. I also re-ran a gel with the sphere to confirm what we got last time. Wei helped us prep samples for the TEM, and he'll image them for us later today (thanks Wei!) since the TEM is booked until 6ish.
- Also changed the color scheme of the wiki. No more crimson!
Week 5
Tuesday (2011-07-05)
Dear Lab Notebook,
- After we left on Friday, Wei imaged the unpurified and purified sphere samples again and found no spheres. Perhaps the folding reaction was not ideal for the formation of spheres; I used the protocol Adam gave me, which asks for 5x staple excess. Han's protocol calls for 10x staple excess. We thought the M13 scaffold was degraded and bad because of the multiple bands on my first gel, but Adam made some origami with it this weekend, so it should be fine.
- More importantly, I also realized that, in the protocol for basic origami folding, by "add 5 uL buffer," "buffer" refers to 10x folding buffer instead of 1x folding buffer. 5 uL in 50 uL is a 10x dilution, after all... welp, that's probably why there were no spheres in the last batch. I was off by a factor of 10. Muy bien Sherrie.
- (Still, strange though that the lanes from the last gel with 1.6 mM MgCl2 had a smear, while those with 8 mM MgCl2 did not...)
- So I remade some 10x Han Buffer 2, added scaffold and the necessary 10x excess staples, and put it all in the thermocycler. I also folded both spherical origami and spheres without equator strands; the latter should be easier to distinguish on the AFM and TEM. Hopefully this time we'll see spheres!
- In other news, a new logo is up. Me gusta.
- In other other news, we're fighting about where the "Literature" link belongs on the front page. This is what happens when you put four anal people together.
Wednesday (2011-07-06)
Dear Lab Notebook,
- Round 2 of sphere ordering is done! Instead of disulfide linkers, I switched our handles and equator staples so they now have photo-cleavable (PC) spacers. Originally I replaced every disulfide link with a PC spacer, but it turns out that PC spacers are maaaaad expensive. So we came up with the idea to alter our lock strand to have the PC spacer; that way, we only need to order one (instead of 18) PC strands for the opening mechanism. The four handles all have a PC spacer, too.
- In the afternoon we ran a gel of the sphere samples.
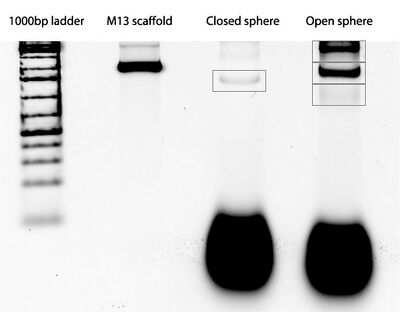
- We also used the AFM and TEM to image the closed and open spheres, and got some really exciting results! The AFM images look really promising.
- Unpurified closed spheres:

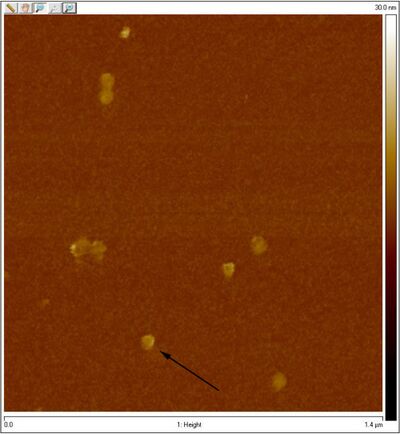
- Unpurified open spheres:
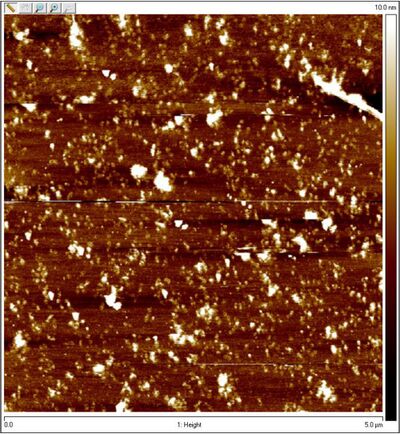
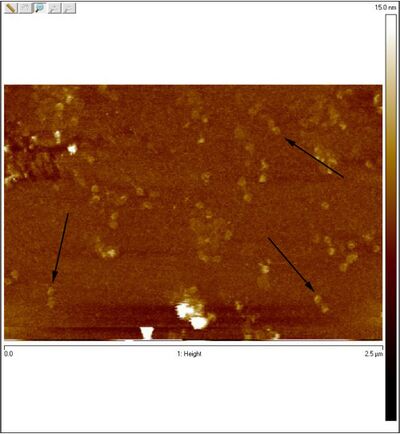
- One of the coolest things is that you can see dimers in the AFM image of open spheres. Perhaps those are the two sphere hemispheres.
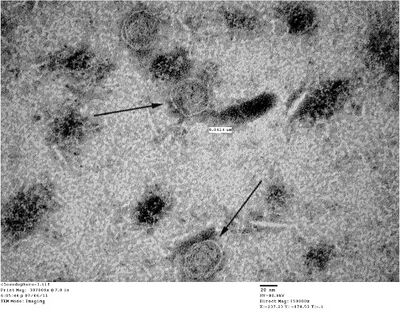
- The TEM images are more ambiguous. Spherical shapes could be water droplets, dirt, etc. But still promising.
- Unpurified closed spheres:
- Unpurified open spheres:
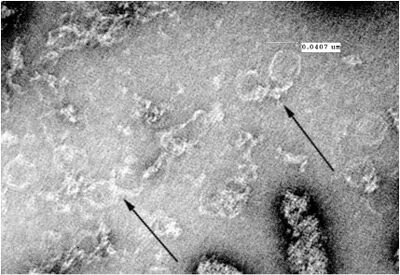







Thursday (2011-07-07)
Dear Lab Notebook,
- Gel purified spheres this morning for imaging. I cut the gel in places indicated in the diagram from yesterday. Also prepped samples for TEM imaging. Unfortunately, the success rate of prepping was rather low... those tiny copper grids are hard to work with.
- Tried AFM on the purified closed sample, but we didn't see much. The TEM was booked this afternoon, so we'll do that tomorrow.
- I also prepared 48-hour reactions for folding the spheres, one closed and one open. Perhaps folding for a longer time will result in better-folded structures that we can see under AFM and TEM.
Friday (2011-07-08)
![]()
Week 6
Monday (2011-07-11)
Dear Lab Notebook,
- I tried to spin column purify 20 uL of the 48-hour spheres this morning, but ended up with next to no product. Measurements with the nanodrop resulted in these measurements:
Sample UV-Vis 260 Reading Purified closed sphere 1 0.005 Purified open sphere 1 0.006 Purified closed sphere 2 0.013 Purified open sphere 2 0.014
- In other words, there's basically no origami in the purification product.
- Later, I ran a gel of the closed and open spheres and got this:
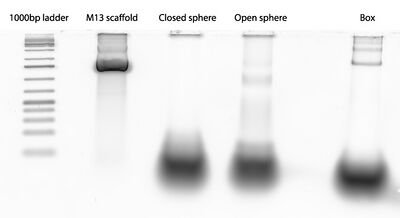
- Where'd the closed spheres go?? Did I forget scaffold or something...
- In the afternoon I redid 12-hour reactions. Tomorrow I want to use AFM to image various open sphere bands from the first sphere gel, and run gels on the new 12-hour sphere reactions.

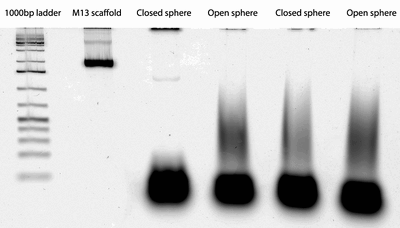
Tuesday (2011-07-12)
Dear Lab Notebook,
- This morning I ran the gel from yesterday's 12-hour folding reaction. There's one faint band in the first closed sphere lane, but I'm not sure what happened with the other three lanes. Perhaps a gel issue?
- In exciting news: the second round of orders came today for the sphere! We now have equator tweezer staples, photo-cleavable handles, and strand displacement handles. The photo-cleavable locks haven't come yet, so I won't be able to fold closed PC spheres until they do. I spent part of the afternoon sorting the new strands into their correct pools.
- I also used the AFM (which I can now operate! hurrah) to image various bands on last week's sphere gel. In the open sphere lane, I imaged the top and middle bands. The top band didn't seem to contain open sphere-like structures, but the middle did.
- I then used the new staples to fold open PC spheres in 12-hour reactions.

Wednesday (2011-07-13)
Dear Lab Notebook,
- To do list:
- Run gel of open PC spheres
- Finish AFMing last week's remaining two samples (open sphere bottom smear, closed sphere) and yesterday's closed sphere band
- AFM raw samples from yesterday
- AFM open PC spheres
- Learn TEM
- TEM open PC spheres
- This morning, I ran a gel of the PC spheres folded for 12 hours. Here's the gel that came out of it:
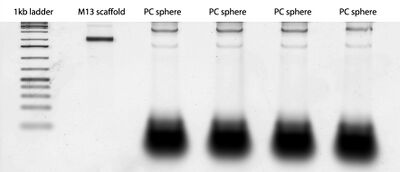
- Throughout the day, I also AFMed last week's open sphere bottom smear and closed sphere samples. I didn't see much for either of them... I don't think gel purification works well for origami.
- AFM for the unpurified PC sphere samples did, however, result in some pretty sweet images:
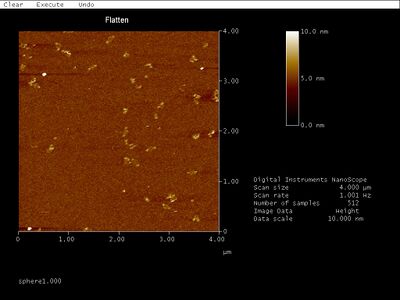
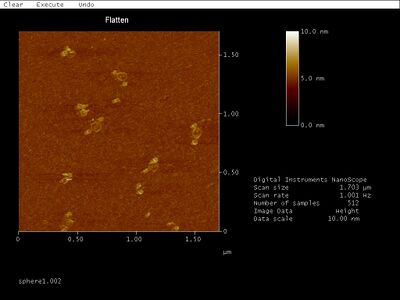
- Just look at the rings between what appear to be two hemispheres! I wonder what they could be. The solid circles are all about 50 nm in diameter, which is close to the theoretical diameter of 42 nm. Given the additional measurement width added by the cantilever tip as well as the flattening of the sphere when subjected to AFM, those solid circles are probably hemispheres. The rings are consistently about 90 nm in diameter. We thought maybe the rings are flattened hemispheres, but a flattened hemisphere should only be 42*pi/2, or 66, nm in diameter. The image also suggests that the inside of the ring is the same height as the mica surface, with the exception of the center of the ring, which is about as raised as the ring itself. Translating this into a side view, I imagine the whole hemisphere-ring complex would look something like this:

- Maybe the ring is created by the middle two rings of the sphere unravelling and unfolding? The diameter would be around 84 nm for such a ring, and that's close to 90 nm. But why would it unravel and consistently become a perfectly circular ring? Doesn't really make sense. And I'm not sure a single helix even shows up on AFM.
- Curious.




Thursday (2011-07-14)
Dear Lab Notebook,
- In the morning, I AFMed the two bands from the PC gel, but didn't seem to see much. I'll repeat this again on Monday.
- In the afternoon, we worked on a presentation for lab meeting. Met with William and Peng, and then gave the presentation at lab meeting.
Friday (2011-07-15)
Dear Lab Notebook,
- Nick, Evan and I attended Jeff Lichtman's 6 hour course on the principles of microscopy today. We thought it would help us understand how AFM and TEM work. Turns out he talked only about optical microscopes imaging on the micrometer scale, which was not so relevant. But still cool to learn about optics and different microscopes; I'm sure someday the knowledge will come in handy.
Week 7
Monday (2011-07-18)
Dear Lab Notebook,
- To Do List:
- Take currently open spheres, add lock strands (to see if two pot folding works)
- Fold closed spheres with strand displacement locks
- Fold closed spheres with PC locks
- Add key / expose to UV light to open closed spheres
- Purify origami
- Image unpurified and purified closed spheres
- This morning, I added lock strands to the PC open spheres in 270-times excess (10-fold excess of one staple is 1*0.05=0.05 uL, but since there are 9 locks per sphere, use 90-fold excess of the strand, which we made 270-fold to be safe, which amounts to 1.35 uL of the lock strand given initial concentration of 100 uM...what a long parenthetical).
- I then tried to purify the spheres using Amicon purification, but again there was no volume in the filter at all after two iterations of filtration. There was, however, stuff left in the filter after the first filtration. So I look the "waste" from the second filtration and spun it through the filter again at 14000 x g for 3:00 and got some stuff left. Imaging this purified sample under the TEM, there looked like there could be open spheres:
- [[Image: TEM image
- AFM images seem to show open spheres consistent with what we've seen before. There is, however, a lot of large gunk that the Amicon filter couldn't remove:
- [[Image: AFM image
Tuesday (2011-07-19)
Dear Lab Notebook,
- To-do list:
- Run gel with:
- ladder
- scaffold
- original closed sphere
- closed SD sphere (folded last night)
- open SD sphere
- open SD sphere w/ lock (overnight)
- purified open SD sphere
- open SD sphere w/ lock and key (1 hour)
- closed SD sphere w/ key (1 hour)
- If 1 hour key isn't enough, tomorrow run gel with:
- key overnight
- key 4 hour
- key 2 hour
- key 1 hour
- AFM and TEM closed spheres
- Purify closed spheres
- Run gel with:
- Today's gel image:
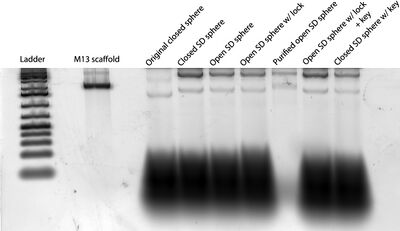
- Conclusions from this gel:
- Amicon purification got rid of excess staples but might have deformed/destroyed open spheres.
- It's hard to tell if there's any difference in the bands between the "Closed SD sphere" and the "Open SD sphere" lanes, so we can't conclude a) if two-pot folding ("Open SD sphere w/ lock") worked or b) if the keys opened the spheres.
- The "Closed SD sphere" may not actually look like the "Original closed sphere." I folded 4 more PCR tubes' worth of Closed SD spheres today and will run another gel tomorrow to confirm/refute these results.
- Also: refolded 4 tubes' worth (200 uL total) of closed spheres again to be gel'd tomorrow.

Wednesday (2011-07-20)
Dear Lab Notebook,
- To-do list:
- Run gel with 1 hour, 2 hour, 4 hour, and overnight key
- Run gel with 4 new closed spheres compared to original closed sphere and open sphere
- AFM & TEM
- Run gel with PC lock (came today!) and lock post-UV exposure
- Gel image of 4 closed sphere lanes is below. I used a 1.5% gel this time instead of 2% in the hopes of getting more separation between closed/open sphere lanes. Seems in fact the same.
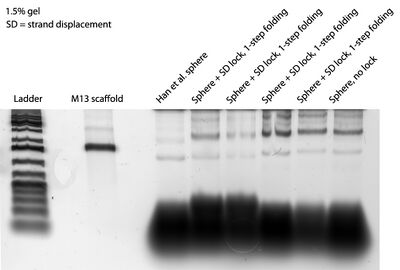

Thursday (2011-07-21)
Dear Lab Notebook,
- Given the gels in the last couple of days, it seems that the following structures may be folding equivalently:
- Sphere + SD lock, 1-step folding
- Sphere + SD lock, 2-step folding
- Sphere without lock (i.e., open sphere)
- This suggests that we're having trouble closing the open sphere using the SD lock.
- Here are some reasons that could explain why this is happening:
- Lack of purification of the scaffold from unbound staples?
- If there are many unbound equator staple handles, then the lock strands we put in may get preferentially bound by these unbound handle strands.
- We've tried to deal with this by using the lock strand in excess of the staples, and we should go further in that direction by adding EVEN MORE lock strands.
- However, we also need to try to get separation of the sphere from unbound staples working by any method. I'll try Amicon purifying the open spheres by spinning only once (twice has historically left no product at the end), and then add lock in various concentrations.
- Too much lock strand?
- One worry is that, if too much lock is being added, then both the top and bottom halves of the sphere will get their own copies of the lock, rather than a single lock strand linking top and bottom.
- To test this, we could do a systematic screen going from around 2x locks relative to the scaffold (the concentration of which we'll get after purification using the nanodrop) to around 45x.
- I'll also give 1-step folding another shot. Instead of adding 270x lock relative to the scaffold, I'll add 171x... the logic for this is the following:
- Say there are X scaffold in solution and 10X of each staple strand.
- Once the scaffold strand is saturated with staples, there are 9X of each staple left.
- Since there are 18 equator staples that have handles that bind to the lock, there are 162X of staples that bind to the lock.
- In order to have ~9X lock left to close the spheres, we should add 171X lock to begin with. We can also screen going from 2X to 45X.
- On the other hand, are we sure that open and closed spheres, once well-formed, do separate on our gels? In the original Han w/ mismatches sphere, with and without the equator, we got good separation. To look into this further, we should do DLS measurements on all of the spheres in our possession.
- If we really can't close an open sphere using these tweezers, then there are other options.
- We can re-design the sphere such that the staples that cross between top and bottom themselves have strand displacement toeholds that can be directly displaced out by 9 different complementary strands. This is not quite as elegant as the idea of using PC spacers at the equatorial junctions, but if it is basically the same. If we can debug the system this way and also get photocleavage working separately, then maybe at the end we can do a version where we actually buy all 9 of the PC crossover staples.
- So. Plan of action moving forward:
Fold 1-step sphere + SD lock with SD lock at even higher concentrations (was at 270x relative to scaffold, increase to 540x <-- maybe even higher?)Fold 1-step sphere + SD lock with 171X SD lockAmicon purify (gently) open SD spheresNanodrop purified open SD spheres- Add lock to purified open SD spheres in 1x, 3x, 9x, 27x, 81x concentrations relative to scaffold
Use DLS to measure hydrodynamic radius of Han et al. original sphere, open Han sphere, closed SD spheres, and open SD spheresRun gel of original closed, original open, SD closed, and SD open spheresAFM closed SD spheres- Re-design sphere as described above
- The gel of the day:
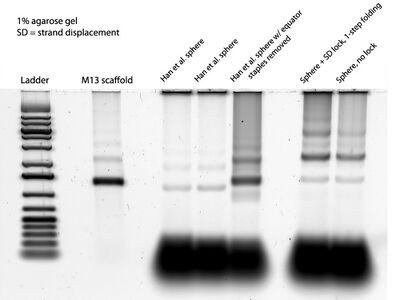

Friday (2011-07-22)
Ahoy there Lab Notebook,
- This morning I ran a gel on the samples I folded last night with different concentrations of lock:
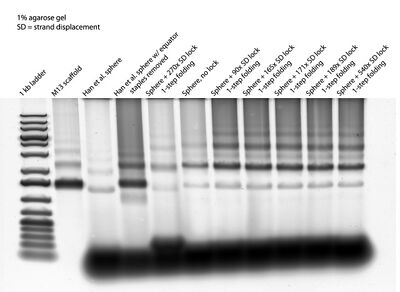
- So, it seems like concentration of lock isn't the (only) reason the lock doesn't seem to be closing the spheres. Nick also used DLS and I imaged the sphere + SD lock with the AFM to confirm gel results. The DLS didn't come out conclusive; particle size by number in a solution with closed/open spherical origami varied over samples, so we're not sure if we can trust the data.
- AFM images of sphere + SD lock:


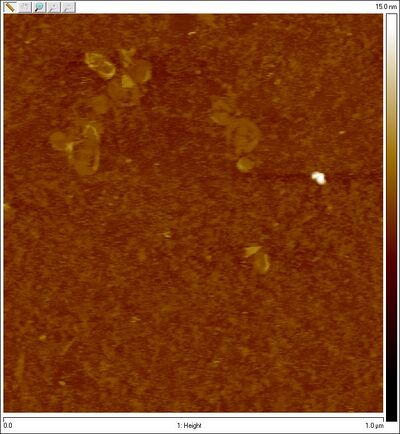
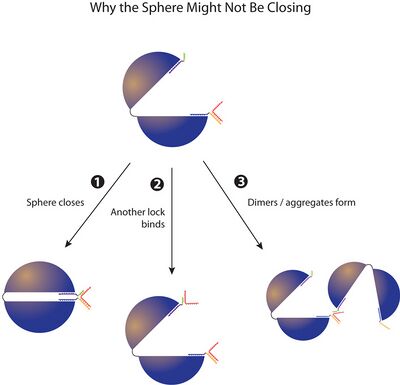
- We now have visual proof that the spheres are not closing (at least, largely open). Time to troubleshoot! I checked Evan's lock sequence to make sure it's complementary to the staples and indeed it is, so that's not the problem. Dave suggests that the problem may be a kinetic one; given 16 bp of hybridization between lock and staple, once the lock and staple bind, they're basically bound forever. The case may be (as suggested previously) that each staple is getting its own lock and the lock is not crossing from the top hemisphere to the bottom one. The locks may also be allowing spheres to form dimers / aggregates.
- Thus, Dave suggests 1) shortening the hybridization sequence to 8 bp to facilitate equilibration after Amicon purification of the open state followed by stoichiometric addition of lock strands, and 2) a cooperative hybridization design with a partial protection strand for the lock (see this paper).
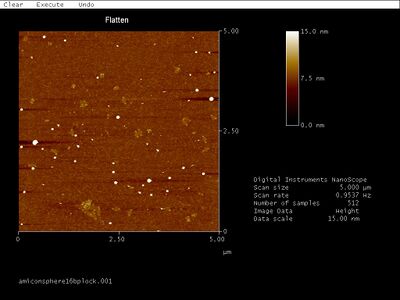
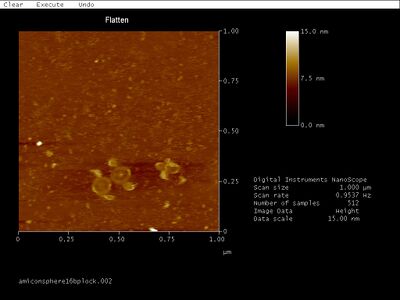
- I also purified the open SD spheres this afternoon and imaged them. This is the first time we're seeing bowl shapes rather than flat circles!!!
- AFM images of Amicon purified open spheres (sphere, no lock):

- (Note the much higher concentration than the previous zoomed-out AFM image!!)
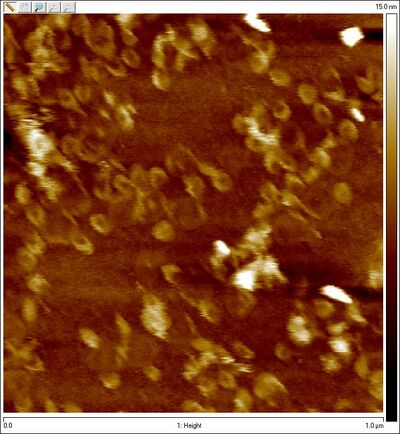
- :)






Week 8
Sunday (2011-07-25)
Dear Lab Notebook,
- There's something that doesn't quite make sense to me about our speculations of why the lock strands aren't closing the spheres...
- If 16-bp hybridization means that a lock strand won't come off once it's bound to a staple, and results in a lock strand becoming bound to each of the 2 sphere staples rather than 1 lock strand binding to both and closing the sphere, doesn't the same problem apply to our usual staple-scaffold interactions, too?
- For example, Staple A is complementary to two different regions A1 and A2 on the scaffold and thereby brings these two regions of the scaffold together. These complementary regions are >=16 bp in length. If 16-bp hybridization is preventing locks from closing the sphere, then it could also prevent a Staple A from bringing together A1 and A2 of the scaffold. That is, Staple A #1 could bind to region A1, and Staple A #2 could bind to region A2. They would each be bound indefinitely, and A1 and A2 would never come together.
- Now, we know origami folding works, so the majority of staples must bring together their scaffold regions a certain percentage of the time. If this is the case for folding origami in general, why isn't it the case for our lock, too? I tried adding the lock at 90x, which is the same concentration we add all our other staples at, so concentration shouldn't be the issue.
- Update: Just had an idea of why high concentrations of lock might result in no spheres closing at all. We've been thinking about the locking process in a very linear way... as in: open sphere folds, one lock binds to its complementary staple in either the Northern or Southern hemisphere, then (as Dave said) there are 3 possibilities from there, 1) sphere closing, 2) another lock binding to the staple on the opposite hemisphere and closing never happening, or 3) dimers/aggregates forming.
- But there's no reason for this to be the sequence of events. In one-step folding, the locks may bind to their complementary staples first, since each is in such high concentration relative to the scaffold. By the time the open spheres are folded, the staples may already be saturated with locks if the lock concentration is higher than the concentration of staples complementary to the lock.
- This may explain why 165x, 171x, 189x, 270x, and 540x lock concentrations aren't closing spheres. But it doesn't explain why the 90x lock concentration spheres didn't fold. Maybe the rate of closing two hemispheres is just very slow.
- Calculations of which interaction, Staple:Scaffold or Staple:Lock, will ultimately win out (courtesy of Dave):
- There are 100 nM of any individual staple and 10 nM of scaffold in solution. The reaction that proceeds is:
- Scaffold + Staple --> Scaffold:Staple
- At Tm1, there are 5 nM of Scaffold:Staple, 5 nM of Scaffold, and 95 nM of Staple.
- [math]\displaystyle{ Keq=\frac{[Scaffold:Staple]}{[Scaffold][Staple]} = \frac{5 nM}{5 nM*95 nM} = \frac{1}{95 nM} = 10^7 / M }[/math]
- There are 100 nM of any individual staple complementary to the lock and 900 nM of lock in solution. The reaction that proceeds is:
- Staple + Lock --> Staple:Lock
- At Tm2, there are 50 nM of Staple:Lock, 50 nM of Staple, and 850 nM of Staple.
- [math]\displaystyle{ Keq=\frac{[Staple:Lock]}{[Staple][Lock]} = \frac{50 nM}{50 nM*850 nM} = \frac{1}{850 nM} = 10^6 / M }[/math]
- At melting temperature Tm,
- [math]\displaystyle{ \Delta G^{\circ}=-R Tm \log{Keq}=\Delta H-Tm \Delta S }[/math]
- [math]\displaystyle{ Tm=\frac{\Delta H^{\circ}}{\Delta S^{\circ}-R \log{Keq}} }[/math]
- Assuming that ΔH° and ΔS° are approximately the same for Scaffold:Staple and Staple:Lock interactions, a larger (more positive) Keq results in a larger numerator in the expression above. Since ΔS° is negative, a larger Keq results in a more negative Tm.
- So, since Keq is larger for the Scaffold:Staple reaction, Tm is lower for the formation of Scaffold:Staple than for Staple:Lock. In other words, when temperature is being lowered in the thermocycler, Staple:Lock forms before Scaffold:Staple.
- Thanks, Dave!
Monday (2011-07-25)
Dear Lab Notebook,
- To-do list:
- Make diagrams of what we're troubleshooting for wiki
Design and order new sphere closing staples, locks, and keysNanodrop and dilute purified open SD spheresAdd lock to purified open SD spheres,wait overnight / varying amounts of timeAdd equator staples to open Han spheres, wait overnight for closing- Run gel tomorrow with purified open SD spheres + lock
- Run gel tomorrow with open Han spheres + equator staples
- Run gel with 90x again (perhaps spheres closed over time)
- I designed new sphere closing staples, locks and keys yesterday and this morning. Here are Nupack results for a couple of the locks, keys, and lock-key complexes.
- Lock and key #1 with high GC content:
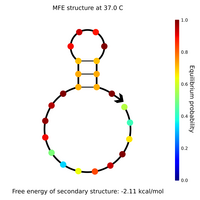
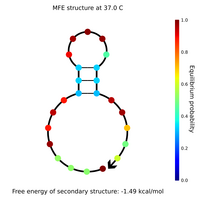
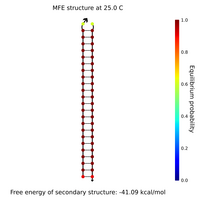
- What Nupack thinks will happen if Lock #1 ("Strand 3") is added in solution with its complementary staples ("Strand 1" and "Strand 2") with all strands at 100 uM:
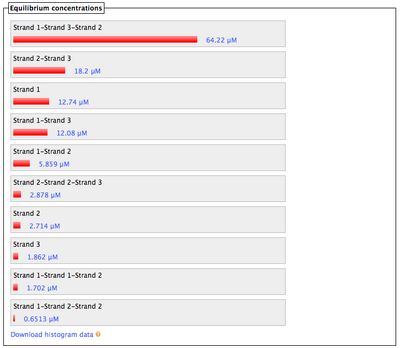
- That looks pretty good; it seems the lock will hold together the two staples.
- What Nupack thinks will happen if Lock #1 ("Strand 3") and Key #1 ("Strand 4") are in solution with lock's complementary staples ("Strand 1" and "Strand 2") with all strands at 100 uM:
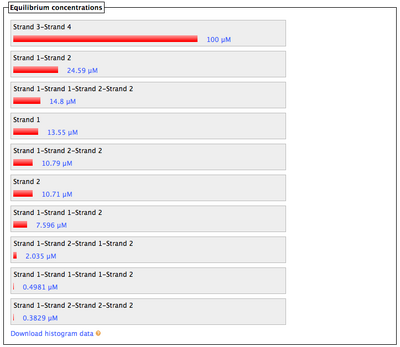
- Also looks good; the lock and key interaction is stronger than all others', meaning the key can displace and free the staples.
- Lock and key #2 with low GC content:
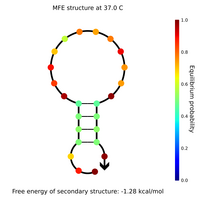
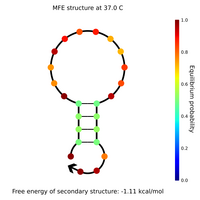
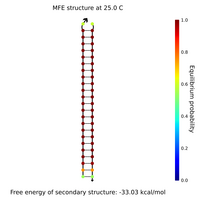








Tuesday (2011-07-26)
Dear Lab Notebook,
- This morning I ran a gel with the purified open SD spheres and locks in various concentrations, as well as the open Han spheres plus equator staples. I forgot to add purified open spheres so I had to run another gel with them (see below). Here's the first gel:
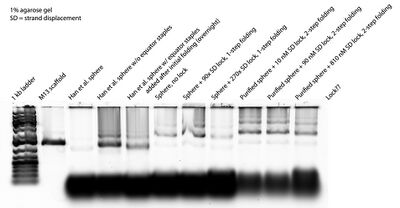
- Things we see:
- Adding equator staples overnight to Han spheres originally folded without equator staples (AKA open spheres) resulted in band shifts toward the closed state (lanes 3-5). This suggests that the equator staples can incorporate themselves into an open sphere and close the sphere.
- "Sphere + 90x SD lock" and "Sphere + 270x SD lock" still look the same as "Sphere, no lock." I understand why this might be the case for 270x, but... what about 90x? There should be no excess lock for 90x.
- Things we don't see:
- I swear I added lock in the last lane, but nothing came up! I'll Nanodrop the lock concentration tomorrow and see what's up.
- Without the purified open spheres, it's impossible to tell if a shift occurred after adding SD lock.
- In the afternoon I ran another gel, with purified open spheres and varying amounts of lock:
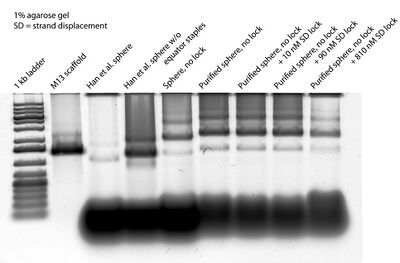
- So... one glaring issue is why the purified open spheres have so much excess staple. There should be no staple... Maybe filtering once wasn't enough?
- Also. It looks like the shift in bands for 10 nM and 90 nM lock are entirely from Amicon "purification." Though there may be a slight "faster" shift in the 810 nM lane... Maybe I should try adding 810 nM SD lock and even higher concentrations and see what happens?
- Lastly, I made a diagram to explain visually why the sphere might not be closing.



Wednesday (2011-07-27)
Dear Lab Notebook,
- To-do list:
- Refine purification of open SD spheres
AFM open SD sphere bands- AFM "open Han sphere + equator staples"
- Add locks to successfully purified open SD spheres
Fold open spheres with handles for Nick
- Dave had an idea today: Since equator staples incorporate into and close open Han spheres, assemble staple-lock-staple complex first, then add to open SD spheres to close them.
- More to-do:
Anneal staple-lock-stapleRun gel with controls- Isolate staple-lock-staple complex
- Add to open SD spheres and run gel
- Our focus until the new locking designs come in for the next couple of days is to try Dave's locking idea, as mentioned above, as well as figure out a good way to purify open spheres. Even though we see a band shift on our gel, the AFM image of purified open SD spheres seems to suggest that after purification we still have open spheres. Perhaps they're flattened or something?
- This morning I AFMed the open SD sphere bands but didn't see anything that looked like open spheres. Granted, there was a lot of noise in the images... Meanwhile, next to me Ralf was making really nice AFM images of rings. I want to be his AFM protege.
- The images are below. Bottom band:
- Close up:
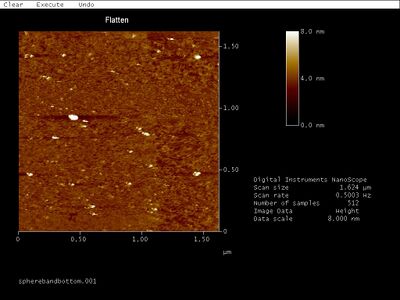
- I saw those small dimer-like spots in an earlier AFM image when I was trying to discern which band was the well-formed structure. Back then, they were about the right size to be open spheres so I concluded they were... But now these same patterns pop up but only ~20 nm wide. So they can't be open spheres. Since they're all oriented the same way, they're might be scanning artifacts.
- Top band:
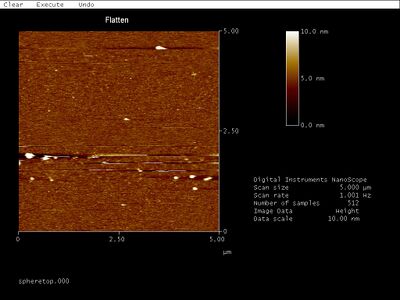
- In conclusion, still not sure which band contains the actual open spheres and which band is another structure / junk.



Donnerstag (2011-07-28)
Liebe Laborjournal,
- Yesterday's gel came out like this (please excuse Evan's red labeling):
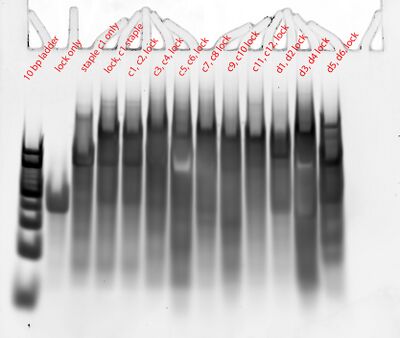
- The weird thing is that there seems to be no change in bands between the "lock + c1 staple" lane and the "lock, c1, c2" lane. The longest oligo length should be ~100 bp for the former and ~160 bp for the latter; that is, a difference of ~60 bp. That should be pretty visible. Nupack also predicts that when lock, c1, and c2 are put in solution in equal concentrations, ~99.76% of the product will be c1:lock:c2. And when lock and c1 are put in solution in equal concentrations, ~100% of the product will be c1:lock. There's no c1:lock:c1. Maybe Evan added c2 to the "lock + c1 staple" sample as well (not to blame it on Evan)?
- In other Nachrichten, I operated the TEM to image a sample for the first time today! Not quite on my own 'cause Adam was there, but close enough. We didn't see much in the sample though; open spheres are hard to make out.
- In the afternoon I remade staple:lock:staple samples and ran a gel. The results look the same as before:
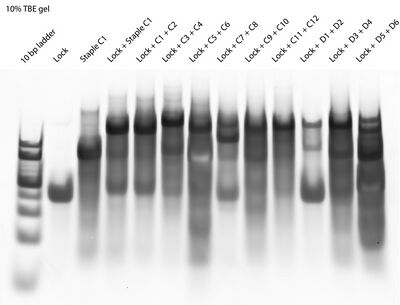
- Still not sure why the "Lock + C1" and "Lock + C1 + C2" lanes are the same. Maybe the C1 well got contaminated with C2? Regardless, we gel purified the dark topmost band in each staple:lock:staple lane. Those bands look ~160 bp, which is the right size. Evan added the staple:lock:staple complexes to unpurified open Han spheres to anneal overnight, so tomorrow we'll hopefully see what we saw when we added equator staples to open Han spheres!
- I also Amicon purified the open SD spheres again. Centrifuging at 14000 x g for 3:50 twice with 500 uL yielded ~100 uL of purified sample in the filter at the end. I'll run a gel tomorrow to see how much staple has been removed. Hopefully my orders will come in too, and I'll be able to fold spheres with the new lock designs.


Friday (2011-07-29)
Dear Lab Notebook,
- To-do list:
Run gel with open Han spheres + staple:lock:staple complexesRun gel with 200 uL open SD spheres, open SD + handle spheres- Gel + Amicon purify open SD spheres
- Run gel to compare gel, Amicon, and gel + Amicon purification methods
- Image purified samples, figure out which band is what
- My IDT order came in today! That means I get to:
Make new pools- Fold 1-step open SD sphere + locks that have 8 bp complementary with equator staples and high GC content (from now on known as "8 bp G locks"; the locks that have 16 bp complementary will be known as "16 bp locks")
- Fold 1-step open SD sphere + locks that have 8 bp complementary with equator staples and low GC content (from now on known as "8bp T locks")
- Fold open SD spheres with 8 bp staples but NO 8 bp lock; this is to be purified later for 2-step sphere closing
- Fold 1-step open Han sphere + equator staples that are complementary to each others (from now on known as "equator staple locks"; not to be confused with "equator staples," which are from the original Han design and have equator crossovers rather than complementary extensions at the equator)
Anneal 20 bp complementary ssDNA strand to 16 bp lock to make cooperative strand displacement lock (from now on known as "cooperative lock")- Gel purify cooperative lock
- Purify open SD spheres and add cooperative lock
- The gel containing open Han spheres + staple:lock:staple complexes is below:
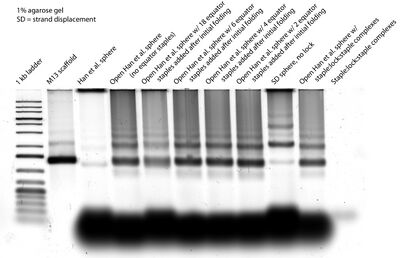
- Dave's idea of incorporating staple:lock:staple strands seems not to have worked because the "Open Han et al. sphere w/ staple:lock:staple complexes" lane looks the same as the "Open Han et al. sphere" lane and different from the "Open Han et al. sphere w/ 18 equator staples added after initial folding" lane. It also seems that adding 2, 4, or 6 equator staples is not enough to close the open Han sphere.
- From what I can tell, closing the sphere with staple:lock:staple complexes can go wrong during annealing of staples and locks, gel purification, or incorporation into the sphere. From yesterday's PAGE gel it seems that some sort of complex annealed; whether it's staple:lock or staple:lock:staple or something else entirely we're not sure. From today's gel (last lane) it looks like the purified sample contained some yield of this complex. Adding this purified complex to open Han spheres resulted in no / an imperceptible change in gel bands. Maybe the annealing reaction didn't result in the desired staple:lock:staple complex (we can try again with much lower than 16 mM [MgCl2])? Maybe the concentration of purified staple:lock:staple (assuming it formed) we added was too low (note how light the band in the last lane is compared to the band in the "Open Han et al. sphere" lane)? Or maybe the staple:lock:staple complexes did not incorporate into the sphere for whatever reason?
- New plan: Adam suggests our goal right now should be to just devise the simplest way possible to close spheres. Right now the most promising method is adding equator staples to open Han spheres. That means the next step is to compile evidence that this method is working, AKA:
- AFM open sphere bands to figure out what band is what structure
- TEM open sphere bands
- AFM open sphere + 18 equator staple band
- This page is getting too long.

Week 9
Sunday (2011-07-31)
- Officially two weeks left! AHHHHHHHHH!!!
- To-do list:
- PRIORITY: Get crystal-clear data (gel and AFM) that adding equator staples to open Han spheres is closing them
- Get really good at gel extraction
- Test new sphere lock designs
- So on Monday:
- Run various % gels on open Han spheres+equator staples to get maximum separation (AKA hog gel boxes)
- Gel purify bands from last Friday's gel of open Han spheres and open Han spheres+equator staples (already cut out)
- AFM various purified bands to figure out which band contains open spheres and to confirm that equator staples closed open spheres
- Fold 8-bp lock spheres, cooperative lock spheres, and equator staple lock spheres
Monday (2011-08-01)
Dear Lab Notebook,
- Ran a 1.5% gel this morning on open Han spheres + equator staples trying to get good band separation and confirm that the equator spheres caused some open Han spheres to close. Separation still doesn't look that clear.
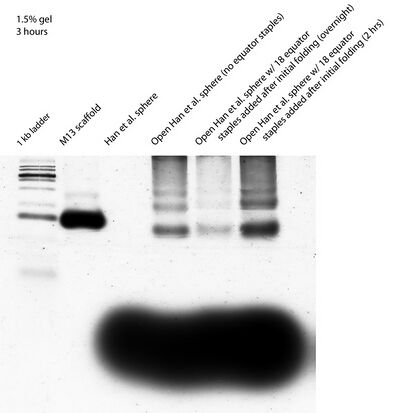
- (The above image was taken the day after the gel was run, so the staples have diffused noticeably through the gel. The usually faint Han et al. sphere band is also no longer visible.)
- Spent the rest of the morning attempting to AFM bands from the open sphere lane, to no avail because the AFM wasn't cooperating. Changed multiple tips, fluid cells, and mica surfaces. Problems were probably due to damaged fluid cells; the springs on a couple are not holding the tips in place well. I'll try again tomorrow since it's important to know which band we ultimately want to look at and compare between lanes, as well as to get more evidence that equator staples are closing open Han spheres.
- In the afternoon, I folded new 1-step spheres with 8 bp G locks, cooperative locks, and equator locks. Couldn't find any more scaffold to do 8 bp T locks or to fold spheres with locks to be added later. The 8 bp G locks and cooperative locks were folded with the locks in 3-fold excess (that is, 27 locks per sphere, where each sphere has 9 lock binding sites).
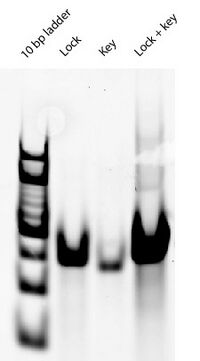
- Also ran a gel to see if last Friday's annealing of the 16 bp lock to its "shield" to make the cooperative lock worked. However, I realized I designed the 20 bp "shield" strand wrong... I meant to alter the key to make it, but instead I just copy-pasted the key and forgot to alter it. Whoops. So the following gel is just annealing the key and lock strands with key in 2x excess. The product looks really light, and there seems to be a lot of unannealed reagent. But we know the key works to displace the lock though based on Evan's min box experiment.


Tuesday (2011-08-02)
Dear Lab Notebook,
- This morning I ran a gel to see if the 8 bp G lock and equator locks close the sphere:
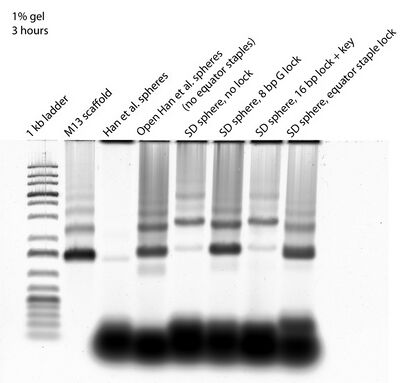
- This looks promising! The lanes with the 8 bp G lock and equator lock look different in terms of band intensity and location compared to the open SD sphere lane. Their bands seem the same as open Han sphere bands, though, which raises suspicion. I don't see how they could be open Han spheres though, given that the parts of the equator staples that bind to the scaffold are identical for the "8 bp G lock" sphere design and "16 bp lock" sphere design. If the "16 bp lock" sphere folded differently from the open Han sphere (which it seems to based on this and past gels), then the "8 bp lock" sphere should also be folding differently from the open Han sphere. Also not sure why intensity of bands is so different...
- Also AFMed a couple of bands from the open SD lanes that Evan excised last week.
- Bottom band:

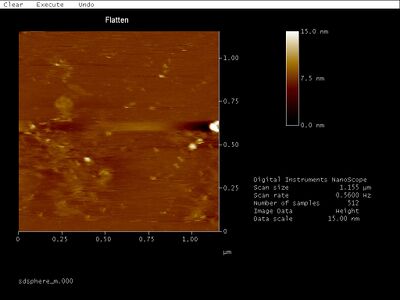
- Could those be open spheres? They're slightly large at around 65-70 nm wide, but that's pretty close to 42 nm considering width added on by the dullness of the tip and/or flattening of the hemispheres. Here's a closeup:

- Middle band:
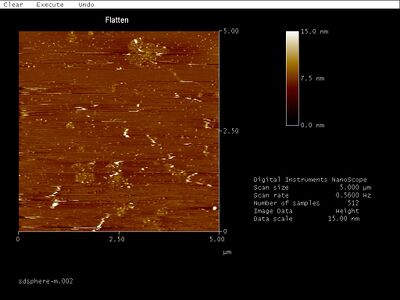
- Looks like a bunch of junk to me... No idea why this even forms a band on the gel.
- Later I ran a 3% gel for 6 hours in hopes of getting better separation, and here's what the gel looks like:
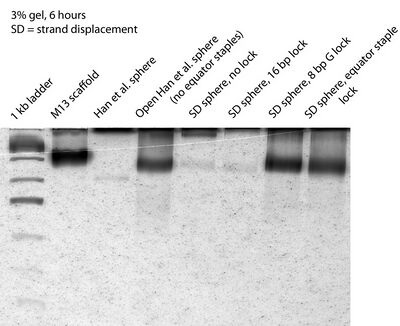
- Not exactly better separation. Will try a 2% gel tomorrow.






Wednesday (2011-08-02)
Dear Lab Notebook,
- To-do list:
Run 2% gel with closed Han sphere, open Han sphere, open Han sphere + equator staples, open SD sphere, open SD sphere + 16 bp lock, open SD sphere + 8 bp G lock, open SD sphere w/ equator lock to get better band separation: want evidence that equator staples close open Han sphere and that 8 bp G lock and equator lock close open SD sphere- AFM open SD sphere top gel band
AFM open Han sphere + equator staples- AFM open Han sphere + staple:lock:staple complexes
AFM open SD sphere + 8 bp G lock- AFM open SD sphere w/ equator lock
Fold open SD spheres w/ equator staples compatible to 8 bp G lock (2-step folding tomorrow)- Add key to open SD spheres + 8 bp G lock ???? <-- if bands shift to open SD sphere, evidence that locks binded and did something
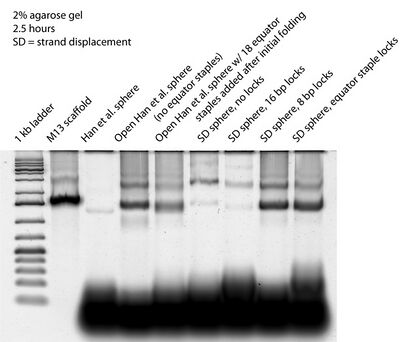
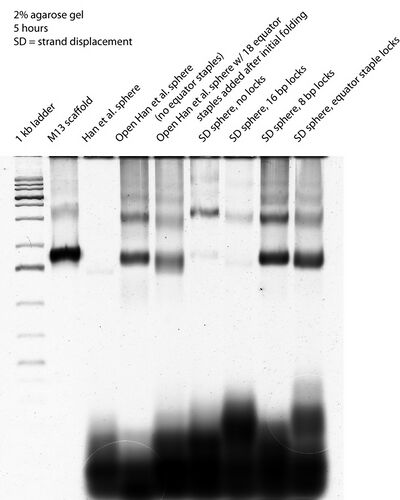
- Interestingly, we see bands shift between the SD sphere (no lock) and the SD sphere w/ 16 bp lock lanes. Previously they had looked the same on gels and we assumed that there was an issue with 16 bp locks closing the sphere. Now it looks like the 16 bp lock may be working; either it worked all along or needed some time sitting around on our lab bench (~2 weeks' worth, since I folded them) to close the spheres.
- In comparison, SD sphere w/ 8 bp locks and equator staple locks have bands that look smeared, and the smears sort of span the space between the bands from the SD sphere (no lock) and SD sphere w/ 16 bp lock lanes. Then again, they still look the same as or very similar to the open Han sphere lane.
- AFM images of open SD sphere + 8 bp G lock:
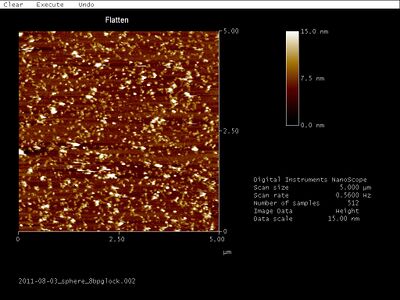
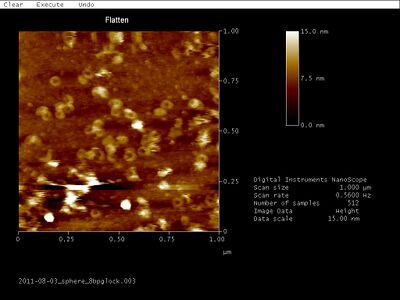
- AFM images of open Han sphere + equator staples:
- INSERT IMAGES
- Purification problem... HPLC, pulse field gel electrophoresis, Pippin Prep, FACS???
- Folded open SD spheres w/ equator staples compatible to 8 bp G lock




Thursday (2011-08-04)
- To-do list:
- AFM open Han sphere + staple:lock:staple complexes
Amicon purify open SD sphere + 8 bp G lockAFM purified open SD sphere + 8 bp G lockAFM open SD sphere w/ equator lockAmicon purify closed and open Han spheresAFM purified closed Han spheres- AFM purified open Han spheres
- Add key to open SD spheres + 8 bp G lock
- Amicon purification
- Amicon-purified closed Han et al. spheres:

- Amicon-purified open SD sphere + 8 bp lock:
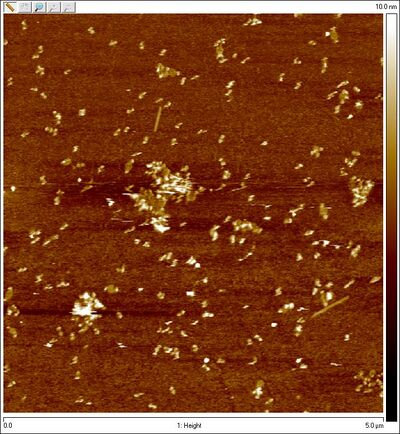
- Unpurified equator lock spheres:

- Could Amicon purification be destroying sphere structures?




Friday (2011-08-05)
Dear Lab Notebook,
- To-do list:
Run gel with more sample for gel extraction of segments of bands/smears- AFM various parts of bands
Run gel to compare unpurified and Amicon-purified samples- AFM Amicon-purified open Han spheres
- AFM unpurified closed Han spheres
AFM Amicon-purified open SD sphere + 16 bp lockTalk to Dave
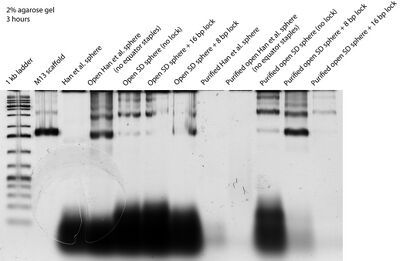
- Today I ran two gels in the morning, one for extracting open Han et al. spheres + equator staples, and one to compare unpurified and Amicon-purified samples to see the effect of Amicon purification on spheres. The gel is below. Unfortunately it came out really bad quality... Evan also ran a gel today and it came out similarly. Maybe the agarose is clumpy or something. I also notice based on the gel that Wei's Amicon filters do a better job of removing excess staples than those other, blue filters that we use (and have now run out of and won't be using anymore).
- Later, I talked to Wei about whether closed spheres would show up on the TEM, and he said they should, since a closed sphere is really 2+ layers of origami. So I tried TEMing closed and open Han et al. spheres again with a sample prep protocol involving double the sample depositing and staining time, but I'm still not sure if we saw spheres or not. There were a lot of circles with ~42 nm diameters that were dark on the inside and outside as expected of origami, but there were also similar circles with ~20, ~70 nm diameters.
- I also talked to Dave over lunch about the kinetics/rate of the 16 bp lock and he said it's possible that it takes a long time for the lock to close the spheres. He suggests incubating the spheres + lock at 45 degrees C to speed up closing. Evan and I also talked to him about why the staple:lock:staple closing didn't work, and why in the control lane with staple A and lock looked the same as the lane with staple A, staple B and lock. He suggested that, due to 1) the nature of DNA synthesis, 2) the fact that the region that binds to the lock on staple B is on the 5' end of a 70-80 bp oligo, and 3) our staples being unpurified, the concentration of staple Bs that have the lock-complementary region may be very low. He further suggested that we add staple B in excess and try the annealing again.
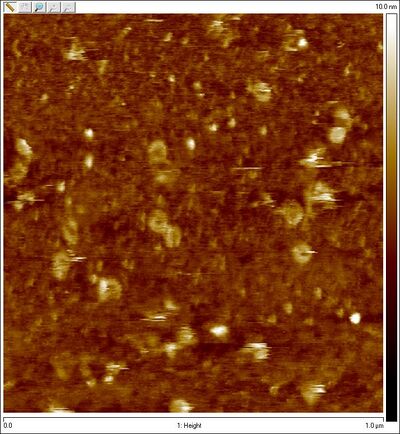
- I AFMed Amicon-purified 16 bp lock spheres, below:
- Though the gel band contains a shift, I see mostly open spheres... Maybe those bright white dots are closed spheres? It's getting frustrating that I don't have a good way to characterize spheres. I'll try gel-then-Amicon purification.
- Lastly, I also added 8 bp lock to open SD spheres in 2-step folding and will run a gel of that on Monday.



Saturday (2011-08-06)
Summary:
- I now have Amicon-purified versions of the following samples:
- Han et al. spheres
- Open Han et al. spheres
- Open SD spheres
- Open SD spheres + 16 bp lock (1-step folding, after 2 weeks)
- Open SD spheres + 8 bp lock (1-step folding)
- And gel-then-Amicon-purified versions of the following:
- Open Han et al. spheres + equator staples, smear top half
- Open Han et al. spheres + equator staples, smear bottom half
- I've AFMed unpurified and purified 1-5 above. So far, I only see what appear to be open spheres. Strangely, when I AFMed the Amicon-purified original Han et al. spheres, I also saw what looked like open spheres, so I'm wondering if Amicon purification destroys the sphere structure. Will investigate this. Still have to AFM open Han et al. spheres + equator staples top and bottom halves of smear.
- I think we've possibly gotten spheres to close with the various locks based on gel evidence, but have yet to confirm with imaging. Important hurdles to overcome are 1) characterizing spheres and 2) purifying spheres. To do so, we can look into a) pulse field gel electrophoresis, b) HPLC, c) Pippin Prep, and d) magnetic beads. (Anything else? Other imaging techniques? Attaching stuff onto spheres to tag them?)
Week 10!!!
Monday (2011-08-08)
Dear Lab Notebook,
- To-do list:
Run gel w/ open SD sphere + 8 bp lock (2-step folding)- AFM open Han et al. spheres + equator staples, top and bottom halves of smear
Re-anneal staple:lock:staple complexes with excesses of staples that bind to lock at 5' end- Run gel of above
Fold more open Han et al. spheres (where'd it all go??)- Add staple:lock:staple to open Han et al. spheres
- Find better way to characterize spheres
- Ran a gel with the open SD spheres + 8 bp locks in a 2-step closing, but the gel was messed up and all the bands came out smudged, lopsided, and broken. It's weird, because I made the gel the same way I've been making them... I'll have to do it again tomorrow.
- In other news, I re-annealed the staple:lock:staple complexes with excesses of staples that bind to the lock at the 5' end and folded more open Han spheres.
Tuesday (2011-08-09)
Dear Lab Notebook,
- To-do list:
Run another gel w/ open SD sphere + 8 bp lock (2-step folding)Run gel of staple:lock:staple complexes- Add staple:lock:staple to open Han et al. spheres
- AFM open Han et al. spheres + equator staples, top and bottom halves of smear
- Find better way(s) to characterize spheres
- Gel with open SD sphere + 8 bp lock in 1- and 2-step folding. Doesn't look like the spheres closed...
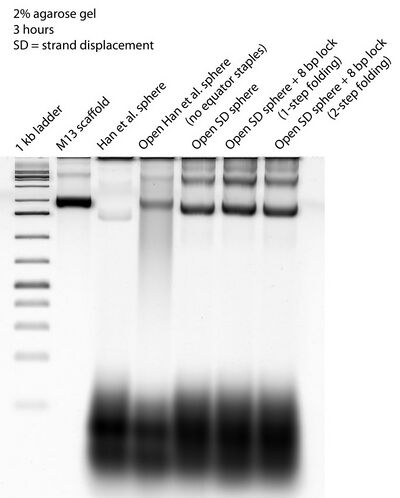

Wednesday (2011-08-10)
Dear Lab Notebook,
- This morning I ran another staple:lock:staple gel (yesterday's lock control lane was messed up). Here it is:
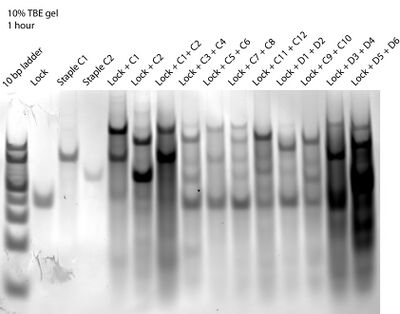
- We followed Dave's suggestion of adding excess staples that bind to lock at the 5' end (we did 8x excess), but note how the lock + C1 + C2 lane still has the same bands as the lock + C1 lane. The only difference I can tell is that the C1 band is darker and the lock + C1 band is lighter in the lock + C1 + C2 lane.
- In the afternoon I tied up some AFM loose ends. I imaged some gel-extracted (but not Amicon-purified) bands from the open SD sphere + 8 bp lock lane. Finally I see spheres! It looks like it's the bottom band that contains open sphere structures.
- Gel-purified open SD sphere + 8 bp lock bottom band:

- Zoomed in:

- Gel-purified open SD sphere + 8 bp lock top band:

- Also AFMed the bottom part of the smear that is open Han spheres + equator staples in search of closed spheres. Here's what I saw:








Thursday (2011-08-11)
Dear Lab Notebook,
- Made our last lab meeting presentation of the summer!
Friday (2011-08-12)
Dear Lab Notebook,
- Last official day of BioMod! :(
- Met with Peng today and said goodbye to Nick and Evan. I'll be back next week, though.
<html>
<a href="http://www2.clustrmaps.com/counter/maps.php?url=http://openwetware.org/wiki/Biomod/2011/Harvard/HarvarDNAnos:LabNotebook_Sherrie" id="clustrMapsLink"><img src="http://www2.clustrmaps.com/counter/index2.php?url=http://openwetware.org/wiki/Biomod/2011/Harvard/HarvarDNAnos:LabNotebook_Sherrie" style="border:0px;" alt="Locations of visitors to this page" title="Locations of visitors to this page" id="clustrMapsImg" />
</a>
</html>

{kind=link}