BioMicroCenter:PPR Program

| HOME -- | SEQUENCING -- | LIBRARY PREP -- | HIGH-THROUGHPUT -- | COMPUTING -- | DATA MANAGEMENT -- | OTHER TECHNOLOGY |
PPR PROGRAM
Guaranteeing high quality next-generation sequencing (NGS) data in a rapidly changing environment is an ongoing challenge. The recent introduction of the Illumina NextSeq500 and the depreciation of specific metrics from Illumina's Sequencing Analysis Viewer (SAV) have made it more difficult to directly determine the baseline error rate of sequencing runs. We have created an open-source tool to construct the Percent Perfect Reads (PPR) plot previously provided by the Illumina sequencers. The PPR program is compatible with HiSeq2000/2500, MiSeq, and NextSeq500 instruments, and provides an alternative to Illumina's Q scores for determining run quality.
The software is designed to be run in a UNIX/LINUX environment.
Dependencies:
Test input fastq file and expected output:
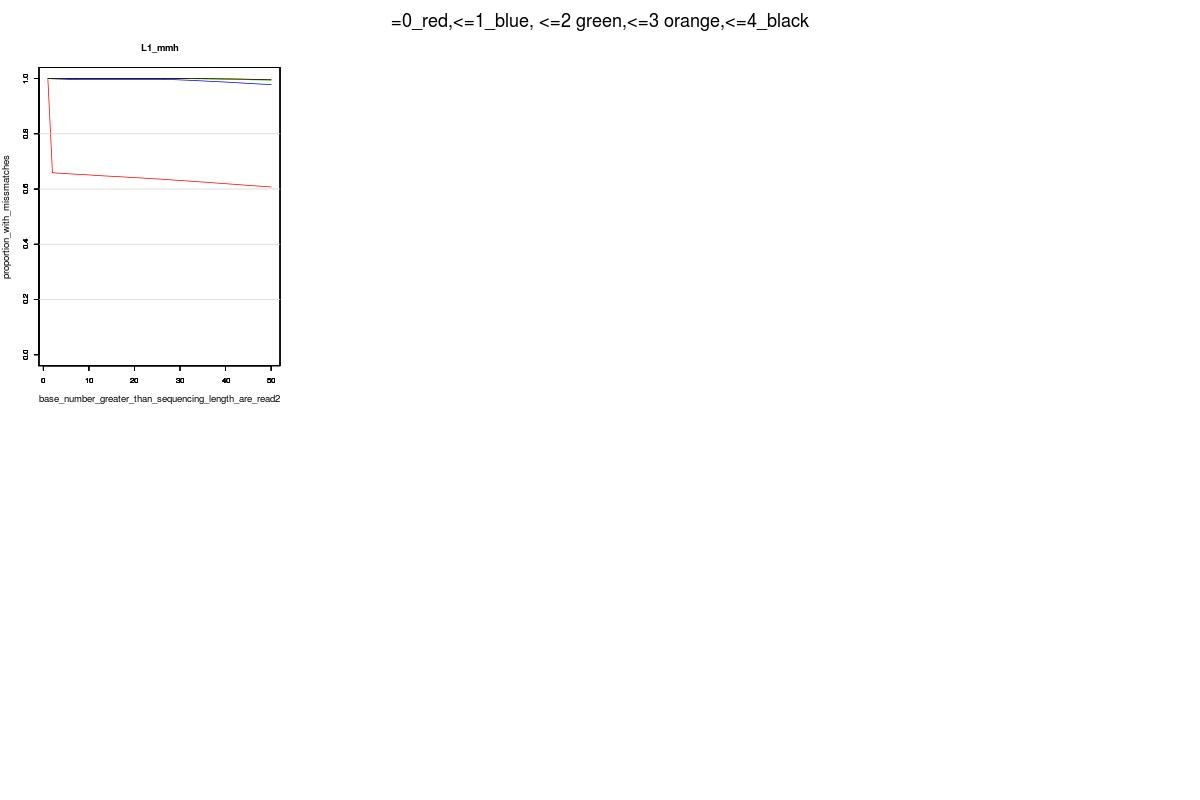
An example Hiseq single end test fastq input file can be downloaded here. This example file is compressed. Make sure to unzip it before your test run. Otherwise, the tool will not work. The expected output basing on the above test file should look like this. If you see the same image, you have all the dependencies installed correctly
Commands:
GENERIC:
cd [CODE DIRECTORY] perl ./NGS_missmatch_qc.pl [RUNTYPE] (ABSOLUTE_PATH/FASTQ FILES)
IMPORTANT NOTE:
The input fastq files cannot be zipped or tarred. The input fastq files must contain absolute path. For example, if the input file read1.fq is under current directory /home/ubunto, the input fastq should be /home/ubunto/read1.fq instead of read1.fq. If the input file read1.fq is under parent directory /home, the input fastq should be /home/read1.fq instead of ../read1.fq. Otherwise, the script will not work.
SPECIFIC:
For Paired end NextSeq sequencing:
./NGS_missmatch_qc.pl nextseq_paired_end absolute_path/read1.fq absolute_path/read2.fq
For single end NextSeq sequencing:
./NGS_missmatch_qc.pl nextseq_single_end absolute_path/read1.fq
For paired end MiSeq sequencing:
./NGS_missmatch_qc.pl miseq_paired_end absolute_path/read1.fq absolute_path/read2.fq
For single end MiSeq sequencing
./NGS_missmatch_qc.pl miseq_single_end absolute_path/read1.fq
For paired end HiSeq sequencing
./NGS_missmatch_qc.pl hiseq_paired_end absolute_path/Lane1_1.fq absolute_path/Lane1_2.fq absolute_path/Lane2_1.fq absolute_path/Lane2_2.fq absolute_path/Lane3_1.fq absolute_path/Lane3_2.fq absolute_path/Lane4_1.fq absolute_path/Lane4_2.fq absolute_path/Lane5_1.fq absolute_path/Lane5_2.fq absolute_path/Lane6_1.fq absolute_path/Lane6_2.fq absolute_path/Lane7_1.fq absolute_path/Lane7_2.fq absolute_path/Lane8_1.fq Lane8_2.fq
For single end HiSeq sequencing
./NGS_missmatch_qc.pl hiseq_single_end absolute_path/Lane1.fq absolute_path/Lane2.fq absolute_path/Lane3.fq absolute_path/Lane4.fq absolute_path/Lane5.fq absolute_path/Lane6.fq absolute_path/Lane7.fq absolute_pathLane8.fq
Results:
See a jpg file named by the time of job submission

{kind=link}