Mssb17/ProtDesign
Computational Enzyme Design: A Practical Exercise
Pablo Carbonell, SYNBIOCHEM Centre, Manchester Institute of Biotechnology, University of Manchester
Objectives
In this exercise, you will learn about bioinformatics tools and databases that can guide you in order to perform an in silico protein design. New tools are continuously becoming available. It is advisable, thus, to check always for the latest developments. For instance, take a look here at some recent publications on the subject.
For this practical exercise, our focus will be on a particular type of protein design problem, the one addressing the challenge of improving catalytic efficiency of an upstream enzyme producing endogenous precursors that are needed for the production of some target compound in E. coli. This problem typically appears if we are interested in compensating the metabolic drain of a precursor essential for growth because of the insertion of the heterologous pathway producing the target compound.
In particular, the objective of this practical exercise is that you learn computational techniques that will allow you:
- To locate functional regions in a protein structure.
- Identifying hot-spots for a specific protein interaction.
- Prediction of best mutations in order to improve the desired protein activity.
Protocol
- Create a model of the transition state of the desired substrate bound to the enzyme
- Position protein side-chain functional groups important for the interaction substrate-enzyme
- Set up a methodology for remodeling the protein backbone when a new side chain is introduced
- Implement a search algorithm+an energy (score) function within the desired constraints
(Adapted from Murphy et al, PNAS, 2009).
Note: we will provide below some illustrative results for a guided example.
Recommended software to install: molecular visualization [1], CHIMERA, Jmol, etc...; format interconversion Open Babel.
Note: check available public Galaxy servers for community-contributed workflows for protein prediction/design.
Choosing the enzyme
Study 1. Select a structure template for the enzyme.
- Select an enzymatic step producing one of the precursors of the biosynthetic pathway of interest, using pathway prediction servers such as XTMS (RetroPath) or metabolic databases like MetaCyc.
- Build a structure template, which will be used during this study to map the predictions.
- Is there a structure of the transition state of the substrate bound to the enzyme?
A selected list of enzymes producing precursors for the targets that you have been studying in the metabolic engineering course can be found in this page.
Note: For all of these enzymes, there are PDB structures available. However, this list is based on annotations that have not been curated. So, it is important to check if actually the structures correspond to enzymes producing the precursors. This information can be checked in KEGG.
If there is no information about the structure, you can search for a template in several databases:
- CSA: Catalyic Site Atlas (link C in RetroPath) in order to get all possible templates.
- Links to 3D structures in UniProt (link U in "RetroPath").
- PDBsum provides comprehensive information about protein structures and their interactions with ligands (you can go from CSA).
If no information is available, still you can analyze this case, but then you will need to build a homology model:
- SWISSMODEL: a fully automated protein structure homology-modeling server.
- CPHmodels 3.0 Server: a protein homology modeling server.
- I-TASSER: provides a hierarchical approach to protein structure and function prediction.
- ROBETTA: full-chain protein structure prediction server.
- BioInfoBank Meta Server(: provides access to various fold recognition, function prediction and local structure prediction methods.
- ESyPred: an automated homology modeling program.
- Geno3D: automated modeler of protein 3D structure
- HHPred: predictor based on HMMs.
- MODELLER: requires local installation and potentially a license.
Guided example: tyrosine aminotransferase
We have selected the enzyme tyrosine aminotransferase in human (EC 2.6.1.5, PDB id [3dyd]):
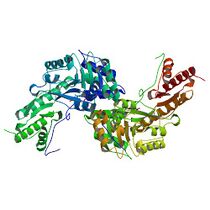
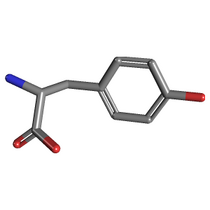
... and its interaction with substrate tyrosine (Pubchem id 6057):


Predicting enzyme active site and locating hot-spots
The prediction of those residues that play a functional role in a protein can be done at different levels, depending on the amount of information available. We will do first predictions based solely on the enzyme sequence or structure, without considering the ligand and the exact location of the catalytic site.
The main criteria here is based on the fact that location of protein interfaces is generally related to some protein features which can be directly measured in the protein structure, such as:
- Sequence conservation
- Amino acid enrichment
- Secondary structure
- Solvent accessibility
- Side chain flexibility
- Side-chain conformational entropy
From single chain structures
Study 2. Find protein functional regions based on descriptors scores.
- Select at least 4 scores from the single-chain algorithms listed below in order to get a residue-wise score of your protein
- Map the highest ranked residues into the 3D structure and visualize the resulting hot regions of the protein.
- How many interfaces does the protein contain?
First of all, we are going to score residues in the protein based on its sequence and/or structural profile.
- Consurf Sequence conservation
- SCORECONS: Score conservation based on multiple sequence alignment.
- SHARP2: Hydrophobic patches.
- PredictProtein: protein prediction suite.
- WHAT IF: Molecular modelling package that is specialized on working with proteins and the molecules in their environment like water, ligands, nucleic acids, etc.
- SABLE: Sequence-based prediction of solvent accessibility; PolyView-2D: Protein structure annotation using sequence profiles.
- cons-PPISP: a method using PSI-Blast sequence profile and solvent accessibility as input to a neural network.
- Promate: a naive Bayesian method based on properties, such as secondary structure, atom distribution, amino-acid pairing and sequence conservation.
- PINUP: a method based on an empirical scoring function consisting of a side-chain energy term, a term proportional to solvent accessible area, and a term accounting for sequence conservation.
- PPI-Pred: a support vector machine method taking six properties (including surface shape and electrostatic potential) as input.
- SPPIDER: a neural-network method that includes predicted solvent accessibility as input.
- Meta-PPISP: a meta web server that is built on raw scores from cons-PPISP, Promate and PINUP through linear regression.
- CASTp: Pockets & cavities
- SurfNet: Surfaces and void regions.
- EvolutionaryTrace: Evolutionary Trace Server.
- ZEBRA: Identifies amino acids responsible for functional diversity based on structural information and physicochemical conservation.
- CAVER: Software tool for protein analysis and visualization to identify tunnels and channels in protein structures.
- FirstGlance: A tool for visualization and analysis of molecular structures.
Docking
Study 3. Build a model of the enzyme-substrate transient state.
- Use a docking tool in order to model the interaction of the substrate with the enzyme.
- Locate those residues that are interacting (less than 8 angstrom) with the ligand and visualize the resulting active region of the enzyme.
- Map the protein active region into the 3D structure and visualize it in the structure of the complex.
- Is there an overlap with the previous identified hot regions?
Web servers:
- Recomended SwissDock: a web service to predict the molecular interactions that may occur between a target protein and a small molecule
- 1-Click Docking: Docking predicts the binding orientation and affinity of a ligand to a target.
- ParDOCK: Automated server for protein ligand docking.
- PatchDock: An automatic server for molecular docking.
Software (needs installation):
- HADDOCK: Protein-ligand docking
- Rosetta
- AutoDock Vina: an open-source program for doing molecular docking.
- Check out many more tools at Click2Drug
Guided example: docking tyrosine aminotransferase
We submit the PDB and the ligand to SwissDock. Since we know that the active site is around Lys280 and the prosthetic group PDP, there is the possibility of performing the docking around this pocket:
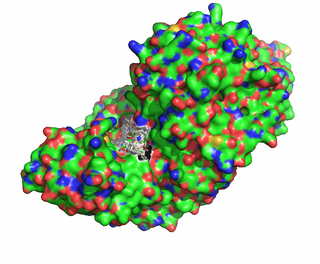
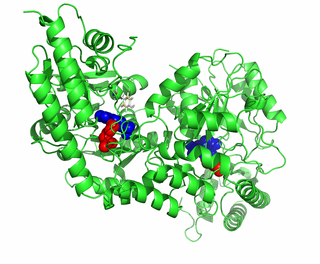
From the ensemble of docked ligands, we select as our transition state model the one with the lowest predicted energy:


Computing the residues at the interface
These are some tools that can provide you the interacting interface of the protein with the small ligand:
- Recommended: Q-SiteFinder: Ligand binding site.
- IBIS: IBIS reports protein-protein, protein-small molecule, protein nucleic acids and protein-ion interactions observed in experimentally-determined structural biological assemblies.
Guided example: substrate interface
We visualize the residues at the interface between the substrate and the enzyme in the model of the transition state:
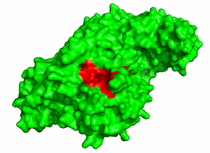

Prediction of hot-spots in the complex structure:
Study 4. Identify enzyme hot-spots.
- Use at least one of the online tools to predict protein hot-spots.
- Map the protein hot-spots, compare their location with the protein interface, and visualize the 3D structure of the complex.
Hot-spots are those residues in the protein that are contributing most to the free binding energy. Experimentally, they are usually determined through an alanine scanning.
- KFC Server]: Protein Interface Hot Spot Prediction.
- Robetta Alanine Scanning: In silico alanine scanning.
- FoldX: Energy-based hot spot prediction tool.
- PredictProtein: Protein prediction suite (ISIS).
- DrugScorePPI: A knowledge-based scoring function for computational alanine-scanning in protein-protein interfaces.
- DrugScore: Score protein-ligand complexes of your interest and to visualize the per-atom score contributions.
- cons-PPISP: Consensus protein-protein interaction interface.
Experimental verification from databases and literature
Study 5. Search for experimentally verified residues.
- Use the experimental databases and literature search in Pubmed in order to find at least one cluster of hot-spots that are functionally related to the interaction.
At this point, it is important to check how close are our predicted hot-spots and active regions to experimentally verified protein residues. There are several databases with experimental data such as point mutations, including alanine scanning data. Furthermore, it is worth to check experimental protein interaction databases to know more about all the interacting partners of the protein, in order to see how can be they related to the protein active regions.
Database of experimental affinities and interface residues
- ASEdb: Alanine scanning database.
- CSA: Catalyic Site Atlas.
- ProTherm: Thermodynamic parameters and wild type of mutant protein.
- WikiBID: Binding Interface Wiki.
- BRENDA: Enzyme information database.
- Ligand Protein DataBase (LPDB): Collection of curated ligand-protein complexes, with 3D structures and experimental binding free energies. Maintained by the university of Michigan.
- AffinDB: Freely accessible database of affinities for protein-ligand complexes from the PDB.
- Protein Ligand Database (PLD): Collection of protein ligand complexes extracted fom the PDB along with biomolecular data, including binding energies, Tanimoto ligand similarity scores and protein sequence similarities of protein-ligand complexes.
- BindingDB: Public, web-accessible database of measured binding affinities, focusing chiefly on the interactions of protein considered to be drug-targets with small, drug-like molecules.
- SCORPIO: Free online repository of protein-ligand complexes which have been structurally resolved and thermodynamically characterised.
- BAPPL complexes set: 161 protein-ligand complexes with experimental and estimated binding free energies calculated with the BAPPL server.
- DNA Drug complex dataset: Dataset of DNA-drug complexes consisting of 16 minimized crystal structures and 34 model-built structures, along with experimental affinities, used to validate PreDDICTA.
- Binding Database: Public, web-accessible database of measured binding affinities, focusing chiefly on the interactions of protein considered to be drug-targets with small, drug-like molecules. Maintained by the Center for Advanced Research in Biotechnology, University of Maryland Biotechnology Institute.
Database of protein interactions
- DIP: Database of Interacting Proteins.
- IntAct: Database system and analysis tools for protein interaction data.
- BOND: The Biomolecular Interaction Network Database (BIND).
- MINT: The Molecular Interactions Database.
- BioGRID: Biological General Repository for Interaction Datasets.
Enzyme Design
Study 6. Combinatorial library of a ranked ensemble of variants.
- Use the [RosettaDesign] webserver in order to perform a //saturation mutagenesis// focused library for the hot-spots positions.
- List the best predicted substitutions for the hot-spots.
- Sketch an in-silico protocol for building a combinatorial library including simultaneous mutations of the hot-spots.
Finally, once the desired positions to mutate have been selected, we perform in silico mutations in order to rank the variants and build a combinatorial library.
Webserver interface
- RosettaDesign: Rosetta design can be used to identify sequences compatible with a given protein backbone. Some of Rosetta design's successes include the design of a novel protein fold, redesign of an existing protein for greater stability, increased binding affinity between two proteins, and the design of novel enzymes.
- Rosie: Rosetta Online Server that Includes Everyone.
Other webservers of interest
- Rosetta VIP: Automated selection of stabilizing mutations in designed and natural proteins.
- PoPMuSiC: tool for the computer-aided design of mutant proteins with controlled stability properties.
- Protein WISDOM: Workbench for in silico de novo design of biomolecules.
- EvoDesign: EvoDesign is an evolutionary profile based approach to de novo protein design.
Stand-alone only
- IPRO: Iterative Protein Redesign and Optimization. IPRO redesigns proteins to increase or give specificity to native or novel substrates and cofactors. This is done by repeatedly randomly perturbing the backbones of the proteins around specified design positions, identifying the lowest energy combination of rotamers, and determining if the new design has a lower binding energy than previous ones. The iterative nature of this process allows IPRO to make additive mutations to the protein sequence that collectively improve the specificity towards the desired substrates and/or cofactors.
- EGAD: A Genetic Algorithm for protein Design. A free, open-source software package for protein design and prediction of mutation effects on protein folding stabilities and binding affinities. EGAD can also consider multiple structures simultaneously for designing specific binding proteins or locking proteins into specific conformational states. In addition to natural protein residues, EGAD can also consider free-moving ligands with or without rotatable bonds. EGAD can be used with single or multiple processors.
- SHARPEN: Systematic Hierarchical Algorithms for Rotamers and Proteins on an Extended Network. SHARPEN offers a variety of combinatorial optimization methods (e.g. Monte Carlo, Simulated Annealing) and can score proteins using the Rosetta all-atom force field or molecular mechanics force fields (OPLSaa).
- Abalone: Software for protein modelling and visualisation.
- Janus: prediction and ranking of mutations required for functional interconversion of enzymes.
Grading
- Study 1. Select a structure template for the enzyme.
- 1.a Select an enzymatic step producing one of the precursors of the biosynthetic pathway of interest
- 1.b Build a structure template, which will be used during this study to map the predictions
- 1.c Is there a structure of the transition state of the substrate bound to the enzyme?
Total: 2
- Study 2. Find protein functional regions based on descriptors scores.
- 2.a Select at least 4 scores from the single-chain algorithms listed below in order to get a residue-wise score of your protein template.
- 2.b Map the highest ranked residues into the 3D structure and visualize the resulting hot regions of the protein.
- 2.c How many interfaces does the protein contain?
Total: 5
- Study 3. Build a model of the enzyme-substrate transient state.
- 3.a Use a docking tool in order to model the interaction of the substrate with the enzyme.
- 3.b Locate those residues that are interacting (less than 8 angstrom) with the ligand and visualize the resulting active region of the enzyme.
- 3.c Map the protein active region into the 3D structure and visualize it in the structure of the complex.
- 3.d Is there an overlap with the previous identified hot regions?
Total: 5
- Study 4. Identify enzyme hot-spots.
- 4.a Use at least one of the online tools to predict protein hot-spots.
- 4.b Map the protein hot-spots, compare their location with the protein interface, and visualize the 3D structure of the complex.
Total: 5
- Study 5. Search for experimentally verified residues.
- 5.a Use the experimental databases and literature search in [Pubmed] in order to find at least one cluster of hot-spots that are functionally related to the interaction.
Total: 3
- Study 6. Combinatorial library of a ranked ensemble of variants.
- 6.a Use the [RosettaDesign] webserver in order to perform a //saturation mutagenesis// focused library for the hot-spots positions.
- 6.b List the best predicted substitutions for the hot-spots.
- 6.c Sketch an in-silico protocol for building a combinatorial library including simultaneous mutations of the hot-spots.
Optional
Updates
Protein design portal
- NewProt portal.
Thermostability
- FireProt: web server for automated design of thermostable proteins.
Solubility
- OU: http://www.biotech.ou.edu/
- SOLPro: http://scratch.proteomics.ics.uci.edu/ (there is a stand-alone version as well)
- PROSO: http://mips.helmholtz-muenchen.de/proso/proso.seam
- ccSOL: http://s.tartaglialab.com/page/ccsol_group
- Protein-Sol: http://protein-sol.manchester.ac.uk
