GRNmap Testing Report 16 Test Files from Dahlquist-data 2015-05-26 TM: Difference between revisions
From OpenWetWare
Jump to navigationJump to search
(add 6 is running) |
(→Results: added 2-genes_47-edges_Dahlquist-data_MM_estimation_fixP-0_no-graph) |
||
| (151 intermediate revisions by 2 users not shown) | |||
| Line 1: | Line 1: | ||
== | ==Test Conditions== | ||
*Date started: 2015-05-26 | |||
* | *Test Performed by: [[User:Tessa A. Morris| Tessa A. Morris]], [[Tessa A. Morris Electronic Lab Notebook| Electronic Notebook]] | ||
*Code Version: [https://github.com/kdahlquist/GRNmap/archive/v1.0.6.zip GRNmap] version 1.0.6 | |||
*MATLAB Version: 2014b | |||
* | *Computer on which the model was run: Row 1 #5, #6; Row 2 #4, #5, #6; Row 3 #1 | ||
* | |||
* | |||
* | |||
''Redone on 2015-06-03 to fix the naming convention and the input sheet'' | |||
*Date started: 2015-06-08 | |||
* | *Test Performed by: [[User:Tessa A. Morris| Tessa A. Morris]], [[Tessa A. Morris Electronic Lab Notebook| Electronic Notebook]] | ||
* | *Code Version: GRNmap-beta (9:17 am 2015-06-08) | ||
*MATLAB Version: 2014b | |||
* | *Computer on which the model was run: Row 2 #4 | ||
* | |||
* | |||
''Update alpha and optimization parameters'' | |||
*Date started: 2015-06-22 | |||
*Test Performed by: [[User:Tessa A. Morris| Tessa A. Morris]], [[Tessa A. Morris Electronic Lab Notebook| Electronic Notebook]] | |||
* | *Code Version: GRNmap-beta (10:28 am 2015-06-22) | ||
* | *MATLAB Version: 2014b | ||
*The computer each input sheet was run on is noted in the results section. | |||
* | |||
* | |||
* | |||
==== | ==Purpose== | ||
*The purpose was to create the sixteen different variations of the input sheet created from dCIN5 Dahlquist-data to test various versions of GRNmap. | |||
* | *[https://github.com/kdahlquist/GRNmap/issues/74#issuecomment-108097323 Issue 74] | ||
* | |||
== | ==Results== | ||
* | *22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-0_fixP-0_graph (Run on Row 2 #4 at 11:51 am 2015-06-22) | ||
== | **[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-0 graph.xlsx]] | ||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-0 graph output.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-0 graph output.mat]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-0 graph plots.zip]] | |||
**[[Media:22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-0_fixP-0_graph_optimization_diagnostic.jpg]] | |||
*22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-0_fixP-0_no-graph (Run on Row 2 #3 at 1:22 pm 2015-06-22) | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-0 no-graph.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-0 no-graph output.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-0 no-graph output.mat]] | |||
**No graph | |||
**[[Media:22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-0_fixP-0_no-graph_optimization_diagnostic.jpg]] | |||
***Had to save manually '''BUG''' | |||
*22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-0_fixP-1_graph (Run on Row 2 #2 at 1:37 pm 2015-06-22) | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-1 graph.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-1 graph output.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-1 graph output.mat]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-1 graph plots.zip]] | |||
**[[Media:22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-0_fixP-1_graph_optimization_diagnostic.jpg]] | |||
*22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-0_fixP-1_no-graph (Run on Row 2 #1 at 1:34 pm 2015-06-22) | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-1 no-graph.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-1 no-graph output.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-1 no-graph output.mat]] | |||
**No graph | |||
**[[Media:22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-0_fixP-1_no-graph_optimization_diagnostic.jpg]] | |||
***Had to save manually '''BUG''' | |||
*22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-1_fixP-0_graph (Run on Row 1 #6 at 1:34 pm 2015-06-22) | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-0 graph.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-0 graph output.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-0 graph output.mat]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-0 graph plots.zip]] | |||
**[[Media:22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-1_fixP-0_graph_optimization_diagnostic.jpg]] | |||
*22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-1_fixP-0_no-graph (Run on Row 1 #5 at 1:45 pm 2015-06-22) | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-0 no-graph.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-0 no-graph output.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-0 no-graph output.mat]] | |||
**[[Media:22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-1_fixP-0_no-graph_optimization_diagnostic.jpg]] | |||
***Had to save manually '''BUG''' | |||
*22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-1_fixP-1_graph (Run on Row 1 #4 at 1:50 pm 2015-06-22 CPU 7) | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-1 graph.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-1 graph output.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-1 graph output.mat]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-1 graph plots.zip]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-1 graph optimization diagnostic.jpg]] | |||
*22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-1_fixP-1_no-graph (Run on Row 1 #4 at 2:07 pm 2015-06-22 CPU 6) | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-1 no-graph.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-1 no-graph output.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-1 no-graph output.mat]] | |||
**No graph | |||
**optimization_diagnostic.jpeg did not save '''BUG''' | |||
*22-genes_47-edges_Dahlquist-data_Sigmoidal_forward_graph (Run on Row 1 #4 at 2:19 pm 2015-06-22 CPU 7) | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal forward graph.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal forward graph output.xlsx]] | |||
**No output .mat file '''BUG''' | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal forward graph plots.zip]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal forward graph optimization diagnostic.jpg]] | |||
**MATLAB error | |||
Error in output (line 140) | |||
outputDiag{3,2} = GRNstruct.GRNOutput.reg_out; | |||
Error in GRNmodel (line 34) | |||
GRNstruct = output(GRNstruct); | |||
:*Repeated and got same error | |||
*22-genes_47-edges_Dahlquist-data_Sigmoidal_forward_no-graph (Run on Row 1 #4 at 2:23 pm 2015-06-22 CPU 7) | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal forward no-graph.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data Sigmoidal forward no-graph output.xlsx]] | |||
**No output .mat file '''BUG''' (''same MATLAB error as above'') | |||
**No graph | |||
**optimization_diagnostic.jpeg did not save '''BUG''' | |||
*22-genes_47-edges_Dahlquist-data_MM_estimation_fixP-1_graph (Run on Row 1 #4 2015-06-22 CPU 7) | |||
**[[Media:22-genes 47-edges Dahlquist-data MM estimation fixP-1 graph.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data MM estimation fixP-1 graph output.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data MM estimation fixP-1 graph output.mat]] | |||
**[[Media:22-genes 47-edges Dahlquist-data MM estimation fixP-1 graph plots.zip]] | |||
**[[Media:22-genes_47-edges_Dahlquist-data_MM_estimation_fixP-1_graph_optimization_diagnostic.jpg]] | |||
*22-genes_47-edges_Dahlquist-data_MM_estimation_fixP-1_no-graph (Run on Row 1 #4 2015-06-22 CPU 6) | |||
**[[Media:22-genes 47-edges Dahlquist-data MM estimation fixP-1 no-graph.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data MM estimation fixP-1 no-graph output.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data MM estimation fixP-1 no-graph output.mat]] | |||
**No graph | |||
**optimization_diagnostic.jpeg did not save '''BUG''' | |||
*22-genes_47-edges_Dahlquist-data_MM_estimation_fixP-0_graph (Run on Row 1 #4 2015-06-22 CPU 5) | |||
**[[Media:22-genes 47-edges Dahlquist-data MM estimation fixP-0 graph.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data MM estimation fixP-0 graph output.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data MM estimation fixP-0 graph output.mat]] | |||
**[[Media:22-genes 47-edges Dahlquist-data MM estimation fixP-0 graph plots.zip]] | |||
**[[Media:22-genes_47-edges_Dahlquist-data_MM_estimation_fixP-0_graph_optimization_diagnostic.jpg]] | |||
*22-genes_47-edges_Dahlquist-data_MM_estimation_fixP-0_no-graph (Run on Row 1 #4 2015-06-22 CPU 4) | |||
**[[Media:22-genes 47-edges Dahlquist-data MM estimation fixP-0 no-graph.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data MM estimation fixP-0 no-graph output.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data MM estimation fixP-0 no-graph output.mat]] | |||
**[[Media:22-genes_47-edges_Dahlquist-data_MM_estimation_fixP-0_no-graph_optimization_diagnostic.jpg]] | |||
***Had to save manually '''BUG''' | |||
*22-genes_47-edges_Dahlquist-data_MM_forward_graph (Run on Row 1 #4 2015-06-22 CPU 3) | |||
**[[Media:22-genes 47-edges Dahlquist-data MM forward graph.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data MM forward graph output.xlsx]] | |||
**No output .mat file '''BUG''' (''same MATLAB error as above'') | |||
**[[Media:22-genes 47-edges Dahlquist-data MM forward graph plots.zip]] | |||
**optimization_diagnostic.jpg did not save correctly. It saved the plot of ACE2 instead. | |||
*22-genes_47-edges_Dahlquist-data_MM_forward_no-graph (Run on Row 1 #4 2015-06-22 CPU 2) | |||
**[[Media:22-genes 47-edges Dahlquist-data MM forward no-graph.xlsx]] | |||
**[[Media:22-genes 47-edges Dahlquist-data MM forward no-graph output.xlsx]] | |||
**No output .mat file '''BUG''' (''same MATLAB error as above'') | |||
**No graph | |||
**optimization_diagnostic.jpeg did not save '''BUG''' | |||
==Discussion== | |||
====Explanation of the intended effect of changing each parameter==== | |||
* Sigmoid: =1 if sigmoidal model, =0 if Michaelis-Menten model | |||
* estimateParams =1 if want to estimate parameters and =0 if the user wants to do just one forward run | |||
* makeGraphs =1 to output graphs; =0 to not output graphs | |||
* fix_P: =1 if the user does not want to estimate the production rate, P, parameter, use initial guess and never change; =0 to estimate | |||
* fix_b: =1 if the user does not want to estimate the b parameter, use initial guess and never change; =0 to estimate | |||
====Observations from test==== | |||
'''May 26, 2015''' | |||
*Setting "makeGraphs" to 1 yielded graphs and when it was set to 0 it did not produce graphs, indicating that the "makeGraphs" command is working properly. | |||
*I did not complete the comparison yet, but so far it looks like there is no change in the production rates for any of the sixteen. | |||
*I only completed comparing the weights between (1) and (2), but they looked identical, which further shows that the "makeGraphs" command is working correctly. | |||
*Tomorrow finish adding in the production rates and comparing the weights. | |||
*Comparing the weights proves to be tedious. The easiest method I found was to copy the matrix and transpose it, copy each column into one master column, create a "master list", sort the weights column by number (smallest to largest), delete the zeros, then sort back into the order of the master list. Then make the list of the Controller ---> target, which is also tedious, however, it does make it easy to double check that the previous step was done correctly. There may be a more efficient way of performing this step. | |||
'''May 27, 2015''' | |||
*[[Media:20150527 GRNmap Test TAR TM.pptx| Preliminary Results]] are shown in the presentation. | |||
*[[Media:20150527 GRNmap Test TAR TM.xlsx| Excel document with analysis]] | |||
*The difference was taken between "Graph" and "No graph" values for the b, production rates, and weights were calculated. Overall there was little to no difference in the values between "Graph" and "No graph." The "Graph" function produced plots each time and the "No graph" did not indicating it was working properly. | |||
*The differences between Sigmoidal and Michaelis Menten could be seen when looking at a bar graph comparing their weights. They seemed to follow the overall shape with the exception of MIG2-->CIN5, MIG2-->MSN2, MSN2-->GCR2, AND MSN2-->HMO1, where there was a change in sign. | |||
**Small magnitude sign changes: ARG80-->MSN2, FKH2-->HMO1, PDR1-->MSN2,SFP1--> MSN2, YHP1-->MSN2 | |||
*It was worth noting that there was primarily positive weights for the Michaelis Menten, with the exception of a few negative weights with very small magnitude ( in between 0 and -0.5) | |||
*When b was fixed (b=1), there was a difference in the network_b values for GLN3 and ZAP1 for the Sigmoidal estimation fixb-1 & fixP-0, Sigmoidal fixb-1 & fixP-1, and Sigmoidal forward only. There was no output for setting b=0, which indicates that fixb-1 is estimating and fixb-0 is fixing b. | |||
*The production rates for "fixP-1" were the same as the production rates in the input sheet, meaning that P was fixed and not estimated, which was the intended function. | |||
*There was no production rate produced for "fixP-0" which should have produced results where the production rates were estimated. | |||
'''May 28, 2015''' | |||
*Format plots of the weight to only show one controller. Make sure that the scale is from -3 to 3 when the min and max and less than the absolute value of 3 and -6 to 6 when not. | |||
*Generate GRNsight maps of each output. Make sure that the nodes are in approximately the same position so the maps can be easily compared. | |||
*[[Media:20150527 GRNmap Test TAR TM.pptx| Final Presentation]] | |||
*[[Media:20150527 GRNmap Test TAR TM.xlsx| Final Excel document]] | |||
*The MM_estimation_fixP-1_graph had no repression (cyan lines) only activation (magenta) and no significance (gray) | |||
*The MM_estimation_fixP-0_graph had four cyan lines, which is still less than all of the Sigmoidal GRNsight maps | |||
*The CIN5 to MIG2 relationship went from MIG2 being strongly repressed (Sigmoid_estimation_fixb-1_fixP-1_graph) to strongly activated (MM_estimation_fixP-0_graph), and was also shown to be insignificant | |||
*The MSN2 to MIG2 relationship: MIG 2 was strongly activated in all models except for the Sigmoidal_estimation_fixb-0_fixP-0_graph_output GRNsight where it was strongly repressed | |||
*HMO1 to MSN2 also changed from MSN2 being repressed (Sigmoidal_estimation_fixb-0_fixP-0_graph, Sigmoidal_estimation_fixb-1_fixP-1_graph, Sigmoid_estimation_fixb-1_fixP-1_graph) to activated (Sigmoidal_estimation_fixb-0_fixP-0_graph). | |||
*GCR2 to MSN2: MSN2 was activated in all cases except Sigmoidal_estimation_fixb-0_fixP-0_graph | |||
[[Category:Dahlquist Lab]] | |||
[[Category: GRNmap]] | |||
Latest revision as of 09:19, 23 June 2015
Test Conditions
- Date started: 2015-05-26
- Test Performed by: Tessa A. Morris, Electronic Notebook
- Code Version: GRNmap version 1.0.6
- MATLAB Version: 2014b
- Computer on which the model was run: Row 1 #5, #6; Row 2 #4, #5, #6; Row 3 #1
Redone on 2015-06-03 to fix the naming convention and the input sheet
- Date started: 2015-06-08
- Test Performed by: Tessa A. Morris, Electronic Notebook
- Code Version: GRNmap-beta (9:17 am 2015-06-08)
- MATLAB Version: 2014b
- Computer on which the model was run: Row 2 #4
Update alpha and optimization parameters
- Date started: 2015-06-22
- Test Performed by: Tessa A. Morris, Electronic Notebook
- Code Version: GRNmap-beta (10:28 am 2015-06-22)
- MATLAB Version: 2014b
- The computer each input sheet was run on is noted in the results section.
Purpose
- The purpose was to create the sixteen different variations of the input sheet created from dCIN5 Dahlquist-data to test various versions of GRNmap.
- Issue 74
Results
- 22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-0_fixP-0_graph (Run on Row 2 #4 at 11:51 am 2015-06-22)
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-0 graph.xlsx
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-0 graph output.xlsx
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-0 graph output.mat
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-0 graph plots.zip
- Media:22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-0_fixP-0_graph_optimization_diagnostic.jpg
- 22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-0_fixP-0_no-graph (Run on Row 2 #3 at 1:22 pm 2015-06-22)
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-0 no-graph.xlsx
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-0 no-graph output.xlsx
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-0 no-graph output.mat
- No graph
- Media:22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-0_fixP-0_no-graph_optimization_diagnostic.jpg
- Had to save manually BUG
- 22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-0_fixP-1_graph (Run on Row 2 #2 at 1:37 pm 2015-06-22)
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-1 graph.xlsx
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-1 graph output.xlsx
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-1 graph output.mat
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-1 graph plots.zip
- Media:22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-0_fixP-1_graph_optimization_diagnostic.jpg
- 22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-0_fixP-1_no-graph (Run on Row 2 #1 at 1:34 pm 2015-06-22)
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-1 no-graph.xlsx
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-1 no-graph output.xlsx
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-0 fixP-1 no-graph output.mat
- No graph
- Media:22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-0_fixP-1_no-graph_optimization_diagnostic.jpg
- Had to save manually BUG
- 22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-1_fixP-0_graph (Run on Row 1 #6 at 1:34 pm 2015-06-22)
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-0 graph.xlsx
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-0 graph output.xlsx
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-0 graph output.mat
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-0 graph plots.zip
- Media:22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-1_fixP-0_graph_optimization_diagnostic.jpg
- 22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-1_fixP-0_no-graph (Run on Row 1 #5 at 1:45 pm 2015-06-22)
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-0 no-graph.xlsx
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-0 no-graph output.xlsx
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-0 no-graph output.mat
- Media:22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-1_fixP-0_no-graph_optimization_diagnostic.jpg
- Had to save manually BUG
- 22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-1_fixP-1_graph (Run on Row 1 #4 at 1:50 pm 2015-06-22 CPU 7)
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-1 graph.xlsx
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-1 graph output.xlsx
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-1 graph output.mat
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-1 graph plots.zip
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-1 graph optimization diagnostic.jpg
- 22-genes_47-edges_Dahlquist-data_Sigmoidal_estimation_fixb-1_fixP-1_no-graph (Run on Row 1 #4 at 2:07 pm 2015-06-22 CPU 6)
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-1 no-graph.xlsx
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-1 no-graph output.xlsx
- Media:22-genes 47-edges Dahlquist-data Sigmoidal estimation fixb-1 fixP-1 no-graph output.mat
- No graph
- optimization_diagnostic.jpeg did not save BUG
- 22-genes_47-edges_Dahlquist-data_Sigmoidal_forward_graph (Run on Row 1 #4 at 2:19 pm 2015-06-22 CPU 7)
- Media:22-genes 47-edges Dahlquist-data Sigmoidal forward graph.xlsx
- Media:22-genes 47-edges Dahlquist-data Sigmoidal forward graph output.xlsx
- No output .mat file BUG
- Media:22-genes 47-edges Dahlquist-data Sigmoidal forward graph plots.zip
- Media:22-genes 47-edges Dahlquist-data Sigmoidal forward graph optimization diagnostic.jpg
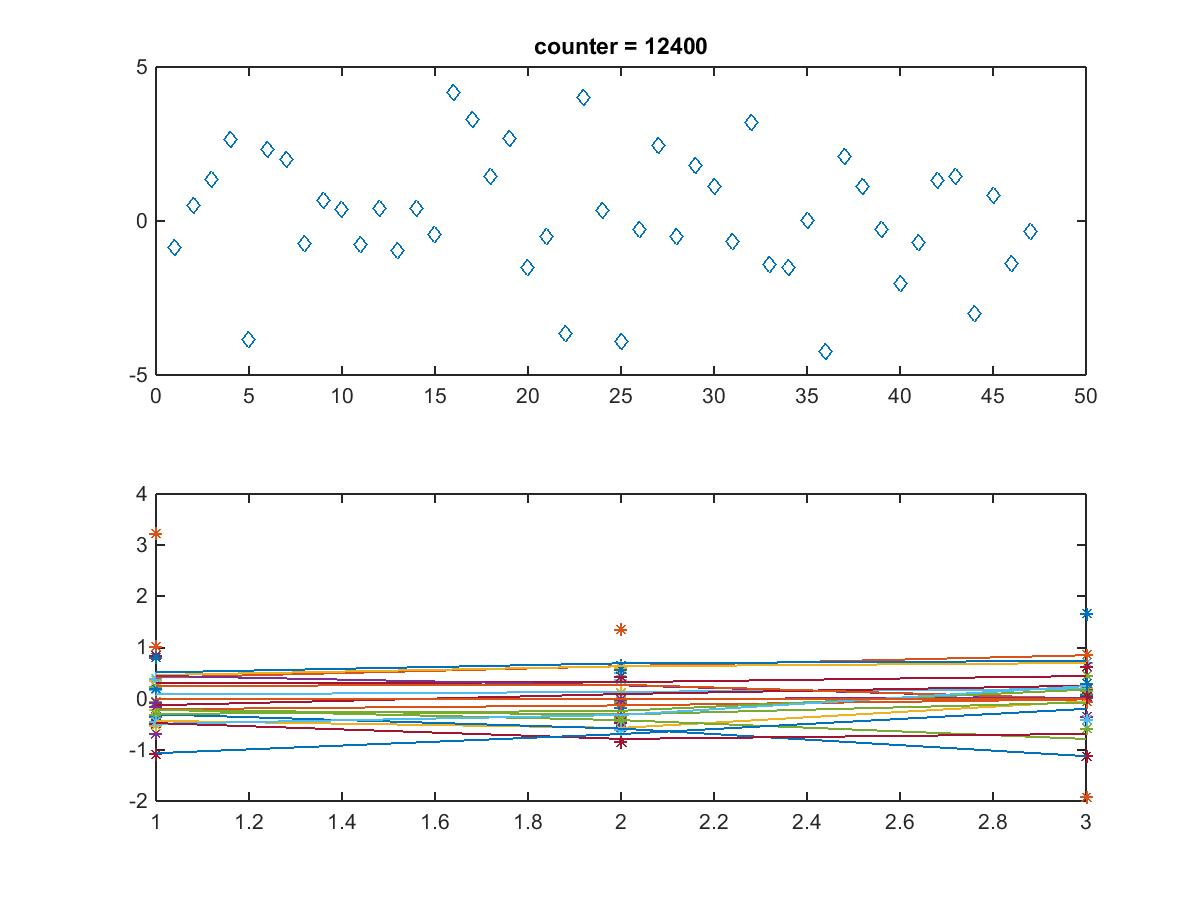
- MATLAB error
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Error in output (line 140)
outputDiag{3,2} = GRNstruct.GRNOutput.reg_out;
Error in GRNmodel (line 34)
GRNstruct = output(GRNstruct);
- Repeated and got same error
- 22-genes_47-edges_Dahlquist-data_Sigmoidal_forward_no-graph (Run on Row 1 #4 at 2:23 pm 2015-06-22 CPU 7)
- Media:22-genes 47-edges Dahlquist-data Sigmoidal forward no-graph.xlsx
- Media:22-genes 47-edges Dahlquist-data Sigmoidal forward no-graph output.xlsx
- No output .mat file BUG (same MATLAB error as above)
- No graph
- optimization_diagnostic.jpeg did not save BUG
- 22-genes_47-edges_Dahlquist-data_MM_estimation_fixP-1_graph (Run on Row 1 #4 2015-06-22 CPU 7)
- Media:22-genes 47-edges Dahlquist-data MM estimation fixP-1 graph.xlsx
- Media:22-genes 47-edges Dahlquist-data MM estimation fixP-1 graph output.xlsx
- Media:22-genes 47-edges Dahlquist-data MM estimation fixP-1 graph output.mat
- Media:22-genes 47-edges Dahlquist-data MM estimation fixP-1 graph plots.zip
- Media:22-genes_47-edges_Dahlquist-data_MM_estimation_fixP-1_graph_optimization_diagnostic.jpg
- 22-genes_47-edges_Dahlquist-data_MM_estimation_fixP-1_no-graph (Run on Row 1 #4 2015-06-22 CPU 6)
- 22-genes_47-edges_Dahlquist-data_MM_estimation_fixP-0_graph (Run on Row 1 #4 2015-06-22 CPU 5)
- Media:22-genes 47-edges Dahlquist-data MM estimation fixP-0 graph.xlsx
- Media:22-genes 47-edges Dahlquist-data MM estimation fixP-0 graph output.xlsx
- Media:22-genes 47-edges Dahlquist-data MM estimation fixP-0 graph output.mat
- Media:22-genes 47-edges Dahlquist-data MM estimation fixP-0 graph plots.zip
- Media:22-genes_47-edges_Dahlquist-data_MM_estimation_fixP-0_graph_optimization_diagnostic.jpg
- 22-genes_47-edges_Dahlquist-data_MM_estimation_fixP-0_no-graph (Run on Row 1 #4 2015-06-22 CPU 4)
- Media:22-genes 47-edges Dahlquist-data MM estimation fixP-0 no-graph.xlsx
- Media:22-genes 47-edges Dahlquist-data MM estimation fixP-0 no-graph output.xlsx
- Media:22-genes 47-edges Dahlquist-data MM estimation fixP-0 no-graph output.mat
- Media:22-genes_47-edges_Dahlquist-data_MM_estimation_fixP-0_no-graph_optimization_diagnostic.jpg
- Had to save manually BUG
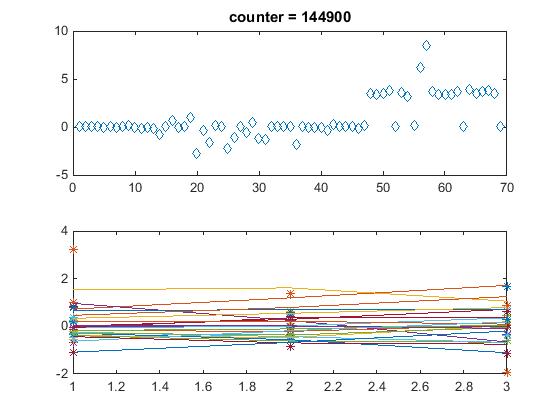
- 22-genes_47-edges_Dahlquist-data_MM_forward_graph (Run on Row 1 #4 2015-06-22 CPU 3)
- Media:22-genes 47-edges Dahlquist-data MM forward graph.xlsx
- Media:22-genes 47-edges Dahlquist-data MM forward graph output.xlsx
- No output .mat file BUG (same MATLAB error as above)
- Media:22-genes 47-edges Dahlquist-data MM forward graph plots.zip
- optimization_diagnostic.jpg did not save correctly. It saved the plot of ACE2 instead.
- 22-genes_47-edges_Dahlquist-data_MM_forward_no-graph (Run on Row 1 #4 2015-06-22 CPU 2)
- Media:22-genes 47-edges Dahlquist-data MM forward no-graph.xlsx
- Media:22-genes 47-edges Dahlquist-data MM forward no-graph output.xlsx
- No output .mat file BUG (same MATLAB error as above)
- No graph
- optimization_diagnostic.jpeg did not save BUG
{kind=link}
{kind=link}
{kind=link}
Discussion
Explanation of the intended effect of changing each parameter
- Sigmoid: =1 if sigmoidal model, =0 if Michaelis-Menten model
- estimateParams =1 if want to estimate parameters and =0 if the user wants to do just one forward run
- makeGraphs =1 to output graphs; =0 to not output graphs
- fix_P: =1 if the user does not want to estimate the production rate, P, parameter, use initial guess and never change; =0 to estimate
- fix_b: =1 if the user does not want to estimate the b parameter, use initial guess and never change; =0 to estimate
Observations from test
May 26, 2015
- Setting "makeGraphs" to 1 yielded graphs and when it was set to 0 it did not produce graphs, indicating that the "makeGraphs" command is working properly.
- I did not complete the comparison yet, but so far it looks like there is no change in the production rates for any of the sixteen.
- I only completed comparing the weights between (1) and (2), but they looked identical, which further shows that the "makeGraphs" command is working correctly.
- Tomorrow finish adding in the production rates and comparing the weights.
- Comparing the weights proves to be tedious. The easiest method I found was to copy the matrix and transpose it, copy each column into one master column, create a "master list", sort the weights column by number (smallest to largest), delete the zeros, then sort back into the order of the master list. Then make the list of the Controller ---> target, which is also tedious, however, it does make it easy to double check that the previous step was done correctly. There may be a more efficient way of performing this step.
May 27, 2015
- Preliminary Results are shown in the presentation.
- Excel document with analysis
- The difference was taken between "Graph" and "No graph" values for the b, production rates, and weights were calculated. Overall there was little to no difference in the values between "Graph" and "No graph." The "Graph" function produced plots each time and the "No graph" did not indicating it was working properly.
- The differences between Sigmoidal and Michaelis Menten could be seen when looking at a bar graph comparing their weights. They seemed to follow the overall shape with the exception of MIG2-->CIN5, MIG2-->MSN2, MSN2-->GCR2, AND MSN2-->HMO1, where there was a change in sign.
- Small magnitude sign changes: ARG80-->MSN2, FKH2-->HMO1, PDR1-->MSN2,SFP1--> MSN2, YHP1-->MSN2
- It was worth noting that there was primarily positive weights for the Michaelis Menten, with the exception of a few negative weights with very small magnitude ( in between 0 and -0.5)
- When b was fixed (b=1), there was a difference in the network_b values for GLN3 and ZAP1 for the Sigmoidal estimation fixb-1 & fixP-0, Sigmoidal fixb-1 & fixP-1, and Sigmoidal forward only. There was no output for setting b=0, which indicates that fixb-1 is estimating and fixb-0 is fixing b.
- The production rates for "fixP-1" were the same as the production rates in the input sheet, meaning that P was fixed and not estimated, which was the intended function.
- There was no production rate produced for "fixP-0" which should have produced results where the production rates were estimated.
May 28, 2015
- Format plots of the weight to only show one controller. Make sure that the scale is from -3 to 3 when the min and max and less than the absolute value of 3 and -6 to 6 when not.
- Generate GRNsight maps of each output. Make sure that the nodes are in approximately the same position so the maps can be easily compared.
- Final Presentation
- Final Excel document
- The MM_estimation_fixP-1_graph had no repression (cyan lines) only activation (magenta) and no significance (gray)
- The MM_estimation_fixP-0_graph had four cyan lines, which is still less than all of the Sigmoidal GRNsight maps
- The CIN5 to MIG2 relationship went from MIG2 being strongly repressed (Sigmoid_estimation_fixb-1_fixP-1_graph) to strongly activated (MM_estimation_fixP-0_graph), and was also shown to be insignificant
- The MSN2 to MIG2 relationship: MIG 2 was strongly activated in all models except for the Sigmoidal_estimation_fixb-0_fixP-0_graph_output GRNsight where it was strongly repressed
- HMO1 to MSN2 also changed from MSN2 being repressed (Sigmoidal_estimation_fixb-0_fixP-0_graph, Sigmoidal_estimation_fixb-1_fixP-1_graph, Sigmoid_estimation_fixb-1_fixP-1_graph) to activated (Sigmoidal_estimation_fixb-0_fixP-0_graph).
- GCR2 to MSN2: MSN2 was activated in all cases except Sigmoidal_estimation_fixb-0_fixP-0_graph